
| Journal of Neurology Research, ISSN 1923-2845 print, 1923-2853 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Neurol Res and Elmer Press Inc |
| Journal website http://www.neurores.org |
Case Report
Volume 1, Number 3, August 2011, pages 109-112
Specific Culomotor Disturbance in a Patient With Amyotrophic Lateral Sclerosis
Naoki Kasahata
Department of Neurology, Makita General Hospital (Division of Neurology, Department of Internal Medicine, Tokyo Metropolitan Ohtsuka Hospital), Minamiohtsuka 2-8-1, Toshima-ku, Tokyo 170-8476, Japan
Manuscript accepted for publication July 28, 2011
Short title: ALS With Specific Oculomotor Disturbances
doi: https://doi.org/10.4021/jnr25w
| Abstract | ▴Top |
A 67-year-old male patient was admitted to the hospital for pneumonia and respiratory insufficiency. Neurological examination showed lower and upper motor system involvement. One year and eight months after the initiation of artificial ventilation, he presented specific oculomotor disturbances. He predominantly showed slow eye movement: saccade speed was approximately 67 degree/second. Sometimes, he showed blink and neck rotation before he initiated saccades, or his saccades were undershoot or did not reach visual objects. He sometimes showed almost normal saccades, especially when he initiated saccades spontaneously (automatically). In this study, we have reported slow eye movement (saccades disturbances) with automatico-voluntary dissociation of an amyotrophic lateral sclerosis (ALS) patient.
Keywords: Amyotrophic lateral sclerosis (ALS); Eye movement
| Introduction | ▴Top |
Till date, the characteristics of oculomotor disturbances of amyotrophic lateral sclerosis (ALS) have not been established. As this character of oculomotor disturbance may be associated with neuropathological character or differential diagnosis of ALS, it seems to be important to make the features of oculomotor disturbances of ALS clear.
Classically, the absence of oculomotor disturbance is the characteristic finding or one of the diagnostic bases of ALS. However, recent studies of ALS patients showed oculomotor disturbances especially after artificial ventilation [1-5]. Slow vertical saccades with rostral interstitial nucleus of the medial longitudinal fasciculus (riMLF) involvement were observed in classical ALS patients with Bunina bodies [6]. Total ophthalmoplegias were observed in ALS patient with multisystem degeneration type [5], superoxide dismutase 1 (SOD1) mutation [4], or basophilic inclusion [5].
However, few publications can be found on the detailed semiology of oculomotor disturbances of ALS.
In this paper, we have reported specific oculomotor disturbances of an ALS patient presented after artificial ventilation.
| Case Report | ▴Top |
A 67-year-old right-handed glazier man was transferred to this hospital because of pneumonia. He had been well until approximately four months before admission, when he began to experience weakness in his bilateral hands and difficulty to perform fine works and open the cap of PET bottle. He was unable to cut glass. Thereafter, he developed difficulty to lift heavy objects, such as glass and bedding (futon). Although he did not develop muscular twitching, he developed diffuse muscle atrophies. Furthermore, he did not develop dysphagia or diplopia. Two months before admission, he developed dysarthria and sputum production. His voice altered from previous one. His body weight had been decreased from 65 to 48 kg. He did not develop any sensory disturbance. Two weeks before admission, his sputum volume increased. He was brought to this hospital in an ambulance by his relatives.
Two years before admission, he suffered from gastric ulcer. There was no apparent family history.
On the second day at hospital, a neurology consultant examined him. The patient showed palmer erythema and did not show any decubitus. Physical examination was otherwise normal.
On neurological examination, the patient was alert and interactive, but showed mild depression and anxiousness during neurological testing. The vital signs were normal. He was oriented to his name, the day, date, month, and year. He knew that he was in a hospital in Tokyo. He was able to recite the months backward, and his speech was fluent with dysarthria. Reading and writing were intact. He recognized his family members without difficulty. The first cranial nerve was not tested; the other cranial nerves and motor, sensory, and coordination examinations were normal, with fasciculation and mild atrophy of tongue, atrophy of intrinsic hand muscles, and mild weakness of bilateral abductor digiti minimi. The patellar and Achilles tendon reflexes were lively bilaterally, while other reflexes were normal. The rest of the examination was normal.
Summary of the neurological examination is as follows: (1) fasciculation and mild atrophy of tongue, (2) distal dominant atrophy of bilateral upper extremities, (3) increased tendon reflexes of bilateral lower extremities and (4) neither apparent ophthalmoparesis nor sensory disturbance.
Laboratory examinations, including urinalysis, showed almost normal findings. Blood examination was normal with WBC 12,900/ul (neutrophil 92.9%), CRP 11.1 mg/dl, and blood glucose 147 mg/dl. Blood gas analysis showed a pH of 7.364, PCO2 of 65.8 mmHg, PO2 of 54.5 mmHg, and HCO3 of 36.7 mmol/l. A chest X-ray showed infiltration shadow of right lower lung field.
On the second hospital day, nasal BIPAP was started. However, his respiratory state was exacerbated. He developed disturbance of consciousness because of respiratory insufficiency. On the third hospital day, he was intubated and ventilated, because he was unable to breathe sufficiently. Thereafter, he became respirator-dependent and tracheostomy was performed on the 16th hospital day. Sometimes, he suffered from pneumonia repeatedly. CT scan showed mild cerebral atrophy and mild dilatation of the III ventricle.
One year and eight months after admission, neurological examination revealed specific ocular movement disturbances. The Bell’s phenomenon was positive. Visual fixations were good without square wave jerk or opsoclonus, and slow pursuits were normal. On examination of reflexive saccades, he predominantly showed slow eye movement or decreased speed of ocular movement. In addition, he sometimes showed blink and neck rotation before he initiated saccades. His saccades were undershoot or did not reach visual objects. Sometimes, he showed almost normal saccades, especially when he initiated saccades spontaneously (automatically) without verbal command. For example, when he saw a physician or a nurse, he showed almost normal saccades. These findings were inconsistent. However, saccades examinations at least for four times consistently showed these oculomotor disturbance findings: slow eye movement, undershoot, and automatico-voluntary dissociation-like phenomenon. Electro-ophthalmogram showed normal pursuits, blinks at saccades initiation, undershoot, or one saccade that did not reach visual objects. The saccades speed was approximately 67 degree/second (Fig. 1).
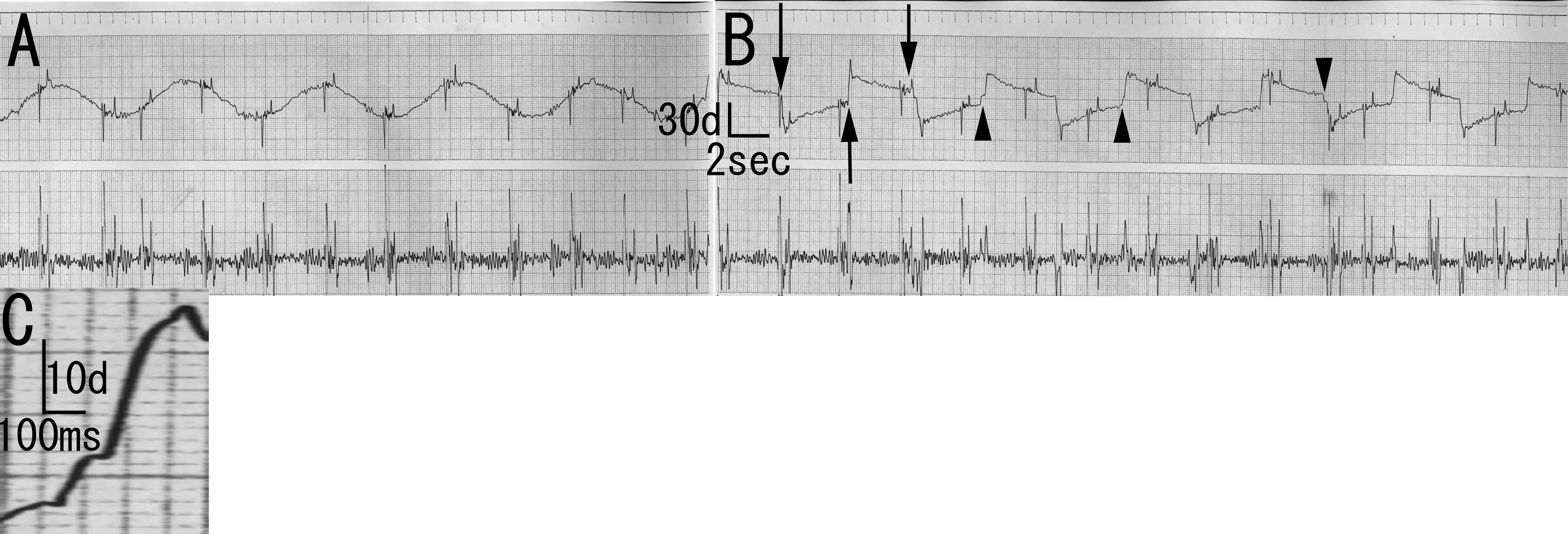 Click for large image | Figure 1. Electro-oculogram of the patient. A: Smooth pursuit. Normal findings. B: Saccades. Blinks are observed at saccades initiation (arrow). There is undershooting (arrowhead). C: Magnification of B. Saccade speed of first part is approximately 33 degree/second. Saccade speed of second part is approximately 67 degree/second. Saccade speed of last part is approximately 150 degree/second. |
| Discussion | ▴Top |
Although ALS patients show oculomotor disturbances after artificial ventilation, detailed clinical semiology has not been established. There are pathological sub-classifications of ALS: classical ALS, multisystem degeneration type, posterior column type (involving SOD1 mutation), and ALS with basophilic inclusion. In addition, SCA1 (or SCA3) sometimes show clinically indistinguishable picture from ALS. Therefore, these disorders have been considered for the differential diagnosis of ALS. ALS patients with long-term course after artificial ventilation show two pathological subtypes: some ALS patients show classical ALS pathology, while others show pathological multisystem degeneration (multisystem degeneration type) [5]. Some familial ALS patients, especially with SOD1 mutation, show posterior column degeneration (posterior column type). Furthermore, juvenile ALS patients as well as some familial ALS patients show basophilic inclusions (ALS with basophilic inclusion) [7-10]. Fused in sarcoma (FUS/TLS) pathology has been reported in ALS with basophilic inclusion [11].
In this study, the ALS patient predominantly presented respiratory insufficiency. He showed mixed lower motor neuron (LMN) and upper motor neuron (UMN) signs: atrophy and fasciculation of tongue and atrophy of intrinsic hand muscle as LMN signs, and increased deep tendon reflexes as UMN signs. After one year and eight months, he presented specific oculomotor disturbances: slow eye movement, blink and neck rotation before initiation of saccades, undershoot, automatico-voluntary dissociation-like phenomenon, and inconsistency.
Only two immediate supra-reticular structures are able to trigger saccades: the frontal eye fields and superior colliculus [12-16]. Experimental and clinical data suggest that the superior colliculus would receive an excitatory afference from the posterior part of the intraparietal sulcus, which could be involved in the triggering of these saccades. The parietal lobe could influence the superior colliculus by increasing the pre-excitation due to the onset of the visual target [15, 16]. It has been proposed that rostral interstitial nucleus of the medial longitudinal fasciculus participates in the production of vertical saccades in man, whereas paramedian pontine reticular formation helps in the production of horizontal saccades [17, 18].
Total ophthalmoplegias were observed in ALS patients with multisystem degeneration [5], SOD1 mutation [4], or basophilic inclusion [5]. However, total ophthalmoplegia was not observed in the patient investigated in our study, whose features were different from previous reports. Total ophthalmoplegia may be a terminal-stage finding of oculomotor disturbance. Slow vertical saccades were observed in classical ALS patients [6]. In addition, slow eye movement was also observed predominantly in the patient included in our study. However, this eye movement was observed at his horizontal saccades. Thus, the absence of total ophthalmoplegia and slow eye movement observed at the patient’s horizontal saccades were different from previous reports. Automatico-voluntary dissociation-like phenomenon (sometimes observed near normal-speed saccades, especially at spontaneous or automatic saccades) and inconsistency suggest cortical origin. Therefore, we presume that specific oculomotor disturbances of the patient might be caused by cortical lesions. However, involvement of the burst neurons of the brainstem may cause specific oculomotor disturbance of the present patient.
However, there was no neuropathological finding of the patient examined in this study. As this patient survived under artificial ventilation, autopsy was not undertaken. Therefore, the problem of the association neuropathological subtype of ALS and these specific oculomotor disturbances remains to be elucidated.
The present ALS patient showed specific oculomotor disturbance one year and eight months after initiation of artificial ventilation. The characteristics of oculomotor disturbances are considered to be associated with the neuropathological subtype of ALS. Specific oculomotor disturbances of this patient may be temporary signs and they may lead to total ophthalmoplegia. In this study, we have reported the slow eye movement (saccades disturbance) with automatico-voluntary dissociation-like phenomenon of an ALS patient.
Grant Support
None.
| References | ▴Top |
- Hayashi H, Kato S. Total manifestations of amyotrophic lateral sclerosis. ALS in the totally locked-in state. J Neurol Sci. 1989;93(1):19-35.
pubmed doi - Hayashi H, Kato S, Oda M. The clinico-pathological findings of the patients grouped as respiratory motor paralysis preceded type of ALS. No To Shinkei. 1999;51(9):771-778.
pubmed - Hayashi H, Oppenheimer EA. ALS patients on TPPV: totally locked-in state, neurologic findings and ethical implications. Neurology. 2003;61(1):135-137.
pubmed - Shimizu T, Kawata A, Kato S, Hayashi M, Takamoto K, Hayashi H, Hirai S, et al. Autonomic failure in ALS with a novel SOD1 gene mutation. Neurology. 2000;54(7):1534-1537.
pubmed - Mizutani T, Sakamaki S, Tsuchiya N, Kamei S, Kohzu H, Horiuchi R, Ida M, et al. Amyotrophic lateral sclerosis with ophthalmoplegia and multisystem degeneration in patients on long-term use of respirators. Acta Neuropathol. 1992;84(4):372-377.
pubmed doi - Averbuch-Heller L, Helmchen C, Horn AK, Leigh RJ, Buttner-Ennerver JA. Slow vertical saccades in motor neuron disease: correlation of structure and function. Ann Neurol. 1998;44(4):641-648.
pubmed doi - Matsumoto S, Kusaka H, Murakami N, Hashizume Y, Okazaki H, Hirano A. Basophilic inclusions in sporadic juvenile amyotrophic lateral sclerosis: an immunocytochemical and ultrastructural study. Acta Neuropathol.1992;83(6):579-583.
pubmed doi - Kusaka H, Matsumoto S, Imai T. Adult-onset motor neuron disease with basophilic intraneuronal inclusion bodies. Clin Neuropathol. 1993;12(4):215-218.
pubmed - Aizawa H, Kimura T, Hashimoto K, Yahara O, Okamoto K, Kikuchi K. Basophilic cytoplasmic inclusions in a case of sporadic juvenile amyotrophic lateral sclerosis. J Neurol Sci. 2000;176(2):109-113.
pubmed doi - Tsuchiya K, Matsunaga T, Aoki M, Haga C, Ooe K, Abe K, Ikeda K, et al. Familial amyotrophic lateral sclerosis with posterior column degeneration and basophilic inclusion bodies: a clinical, genetic and pathological study. Clin Neuropathol. 2001;20(2):53-59.
pubmed - Munoz DG, Neumann M, Kusaka H, Yokota O, Ishihara K, Terada S, Kuroda S, et al. FUS pathology in basophilic inclusion body disease. Acta Neuropathol. 2009;118(5):617-627.
pubmed doi - Hirayama K. Neurological Semeiology (Japanese), revised 2nd ed., vol. 1 2006:546-551.
- Pierrot-Deseilligny C. Central oculomotor circuits. Rev Neurol. 1985;141(5):349-370.
pubmed - Vighetto A, Perenin MT. Optic ataxia: analysis of eye and hand responses in pointing at visual targets (author's transl). Rev Neurol. 1981;137(5):357-372.
pubmed - Pierrot-Deseilligny C. Cortical control of saccades. Rev Neurol. 1989;145(8-9):596-604.
pubmed - Pierrot-Deseilligny C, Gray F, Brunet P. Infarcts of both inferior parietal lobules with impairment of visually guided eye movements, peripheral visual inattention and optic ataxia. Brain. 1986;109(Pt 1):81-97.
pubmed - Buttner-Ennever JA, Buttner U, Cohen B, Baumgartner G. Vertical glaze paralysis and the rostral interstitial nucleus of the medial longitudinal fasciculus. Brain. 1982;105(Pt 1):125-149.
pubmed - Buttner-Ennever JA, Buttner U. A cell group associated with vertical eye movements in the rostral mesencephalic reticular formation of the monkey. Brain Res. 1978;151(1):31-47.
pubmed doi
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Neurology Research is published by Elmer Press Inc.