Figures
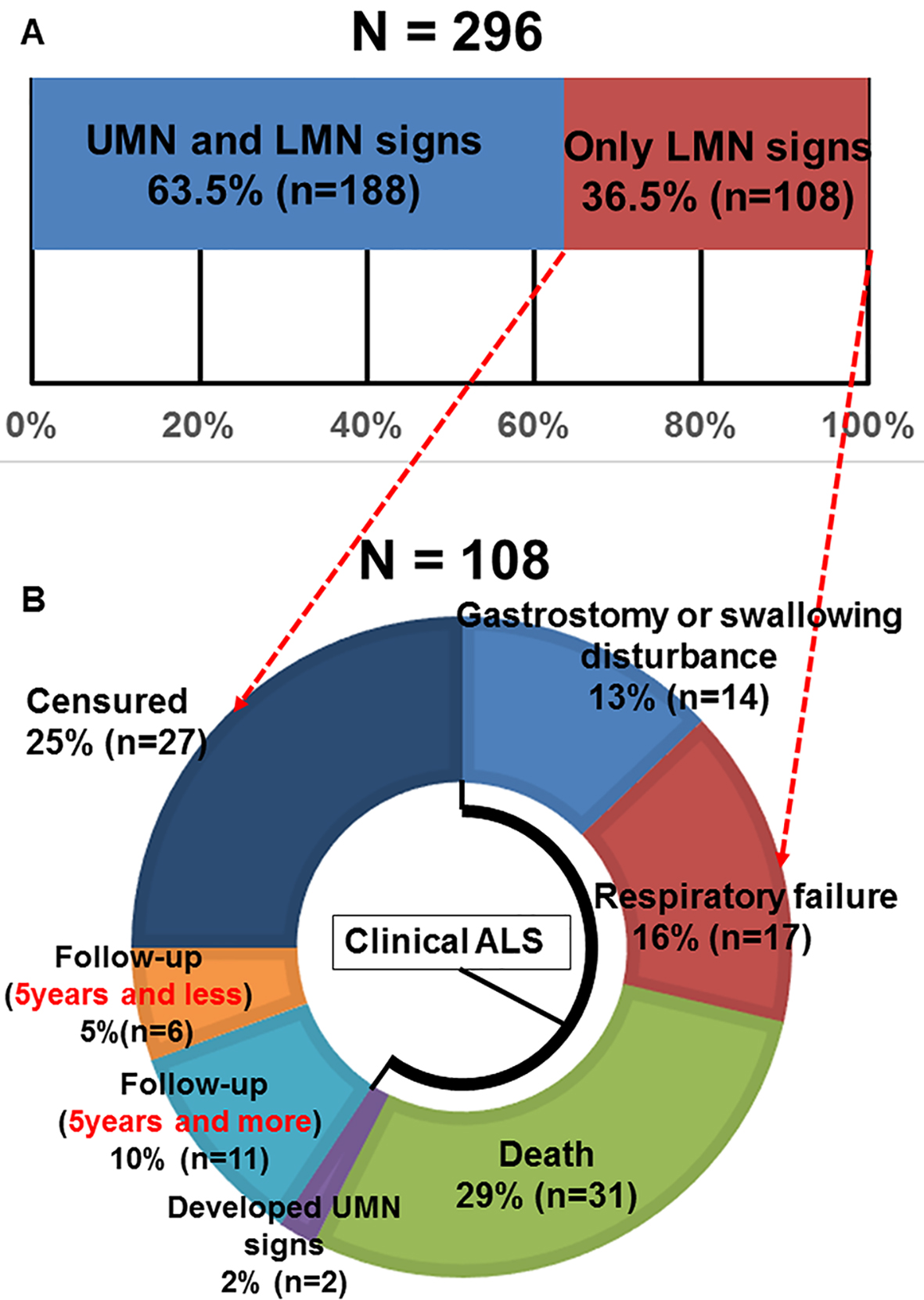
Figure 1. (A) Motor neuron disease patient’s percentages according to motor neuron signs (UMN: upper motor neuron; LMN: lower motor neuron). (B) The outcome of 108 cases whose definite, probable and possible ALS diagnosis was unable to be confirmed due to only LMN signs during follow-up periods.
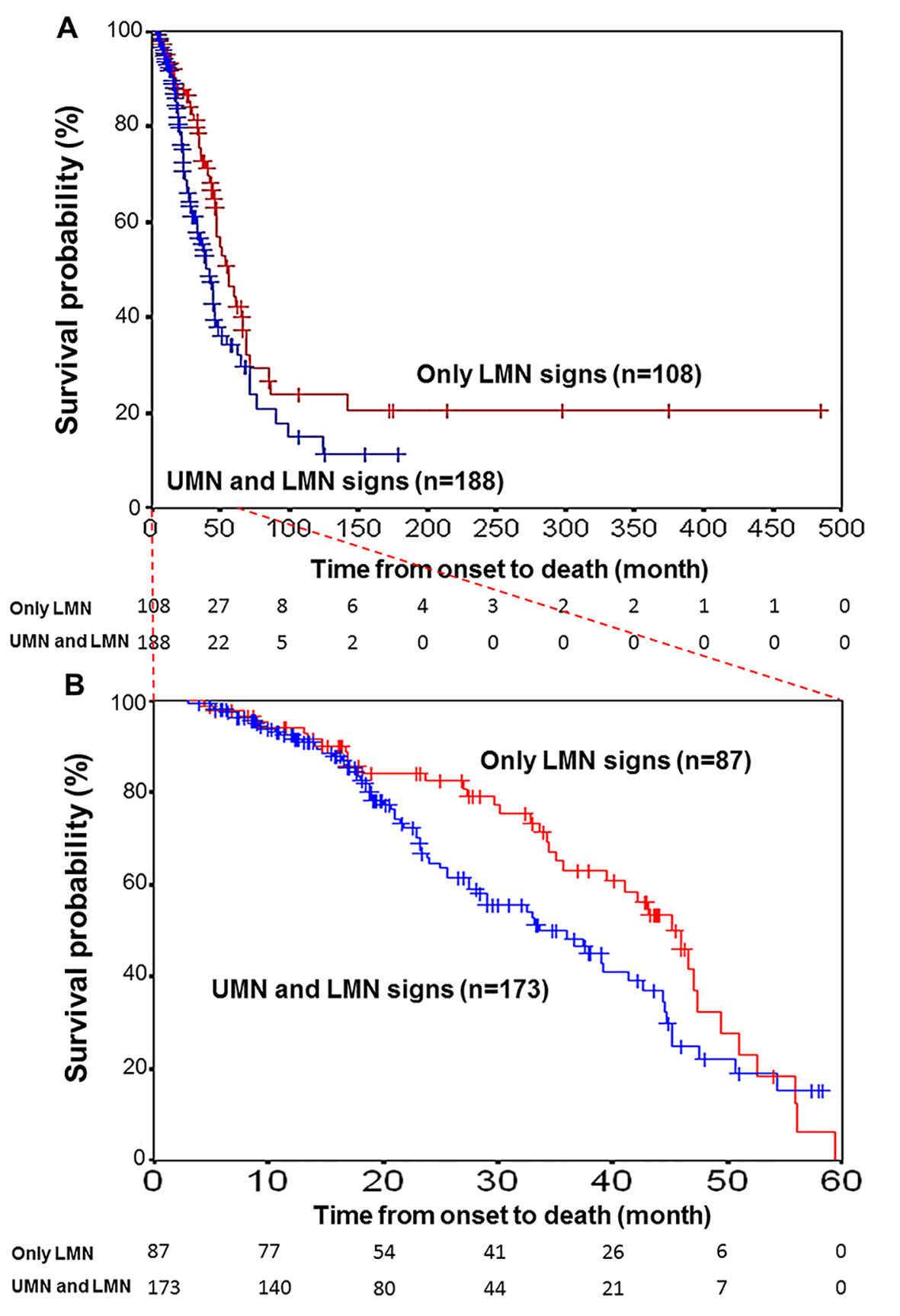
Figure 2. Kaplan-Meier survival curves for patients with motor neuron disease. (A) Kaplan-Meier survival curves for motor neuron disease patients (N = 188) with UMN and LMN signs (blue line) versus those (N = 108) with only LMN signs (red line) for all 296 patients according to the motor neuron symptoms status. Vertical bars indicate censured data. A significant difference was found between the two survival curves (log-rank test, P < 0.05). (B) Time from onset to death was limited to 5 years by examination. The curves of 260 patients (173 with both UMN and LMN signs and 87 with only LMN signs) with vs. without UMN signs did not reveal any difference (log-rank test, P = 0.06).
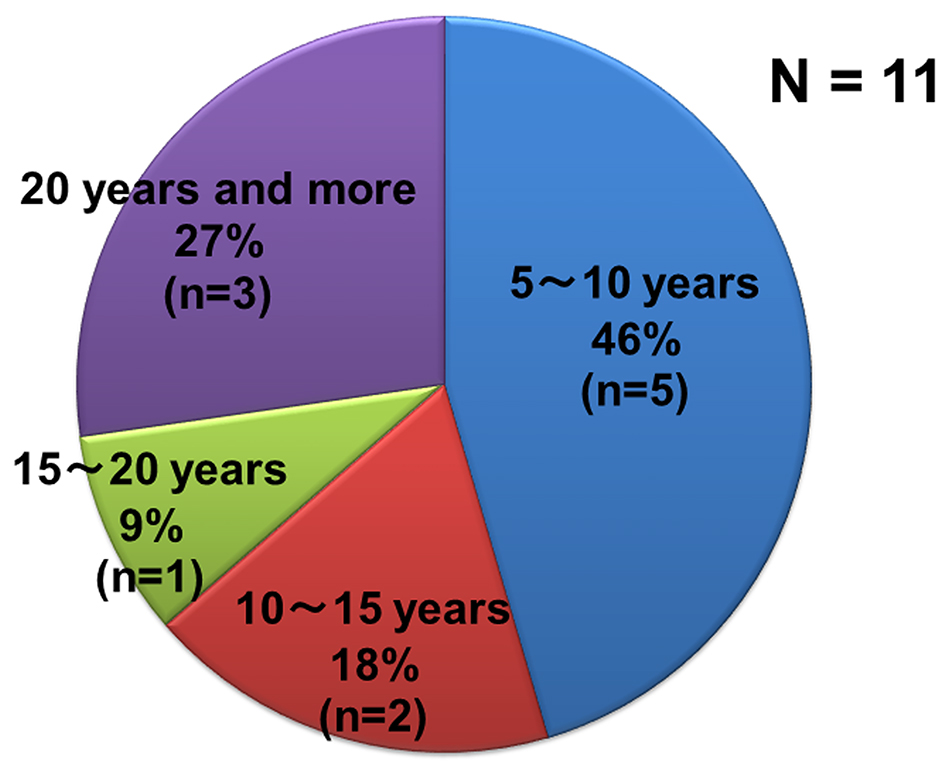
Figure 3. Percentage of disease durations of patients with only LMN signs and who have been not diagnosed as ALS.
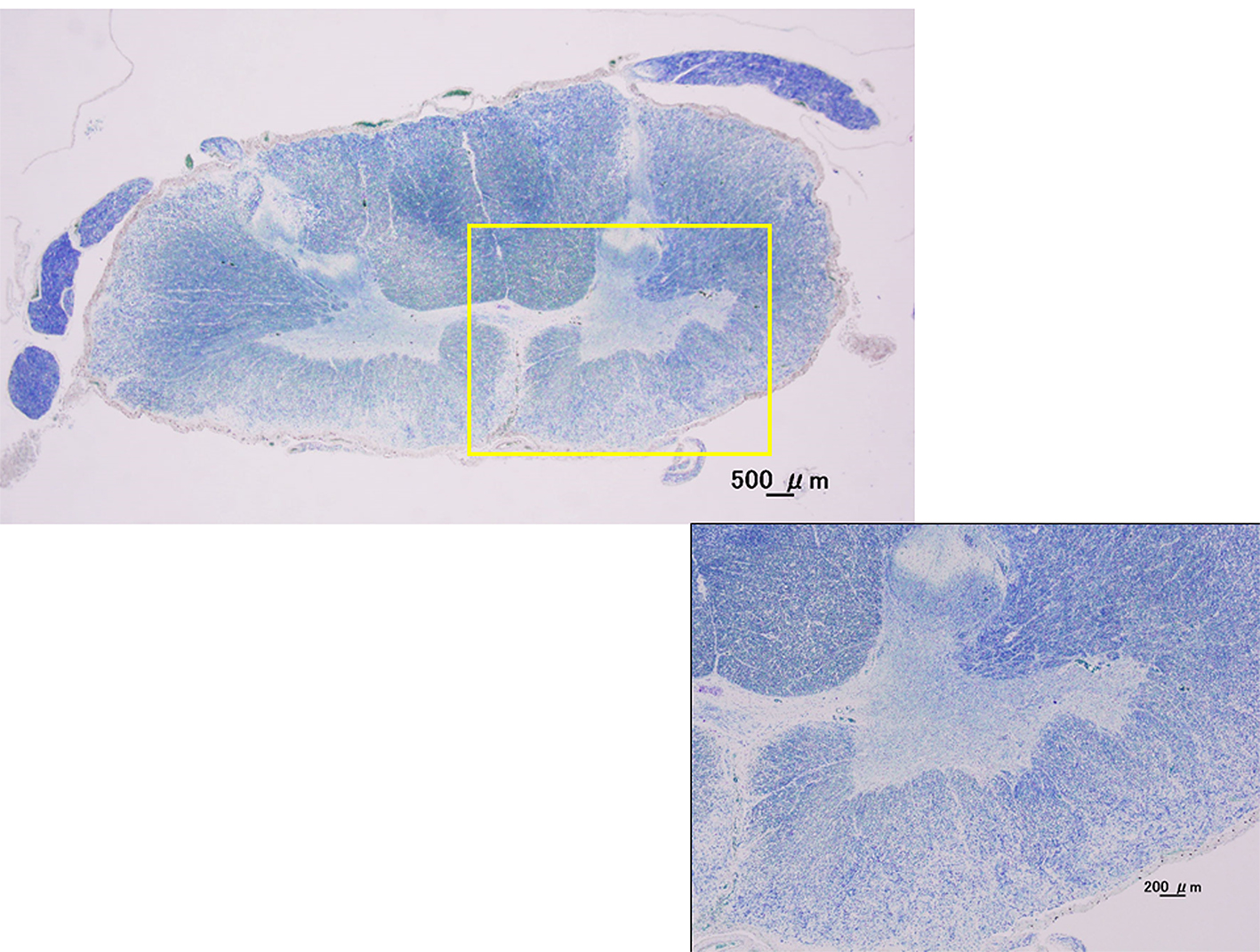
Figure 4. Kluver-Barrera stain. Cervical spinal cord (C7). Histological examination showed not only atrophy of the anterior horns of the spinal cord, but also the extensive dropout of anterior horn cells in cervical spinal cord with the pure LMN cases by autopsies.
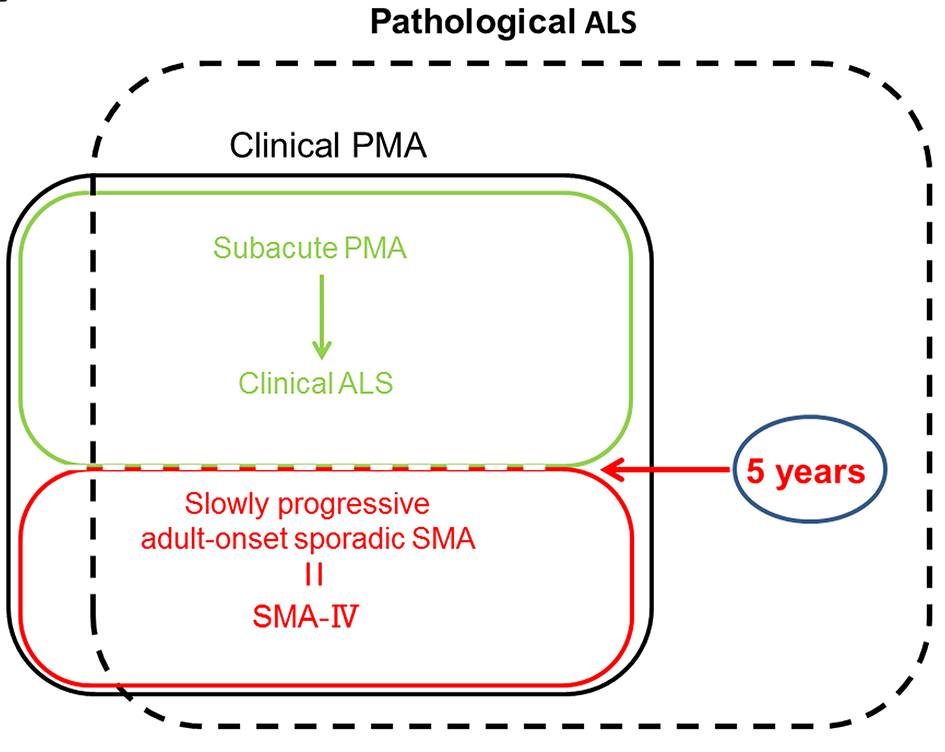
Figure 5. The medical positioning between ALS, PMA and adult-onset sporadic SMA. PMA: progressive muscular atrophy; SMA: spinal muscular atrophy.
Tables
Table 1. ALS Phenotypes as Recognized by Clinicians
| Type | Characteristic |
|---|
| ALS: amyotrophic lateral sclerosis; PMA: progressive muscular atrophy; UMN: upper motor neuron; LMN: lower motor neuron. |
| ALS | Mixed UMN and LMN sings in limbs and bulbar region |
| PMA | Pure LMN syndrome, generally asymmetrical |
| Primary lateral sclerosis | Pure UMN syndrome |
| Progressive bulbar palsy | Isolated UMN or LMN signs, or both, confined to bulbar musculature |
| Flail arm syndrome | Symmetrical, proximal, largely LMN weakness of upper limbs |
| Flail leg syndrome | Symmetrical, distal, largely LMN weakness of lower limbs |
Table 2. Baseline Characteristics of Patients With Those Motor Neuron Disease
| Characteristic | UMN and LMN signs | Only LMN signs | P value |
|---|
| UMN: upper motor neuron; LMN: lower motor neuron. |
| The mean age at onset | 61.8 ± 11.2 | 62.3 ± 10.7 | t-test, P = 0.76 |
| Sex (male/female) | 100/88 | 58/50 | χ2 test, P = 0.93 |
| Bulbar symptom (+/-) | 116/72 | 39/69 | χ2 test, P < 0.05 |