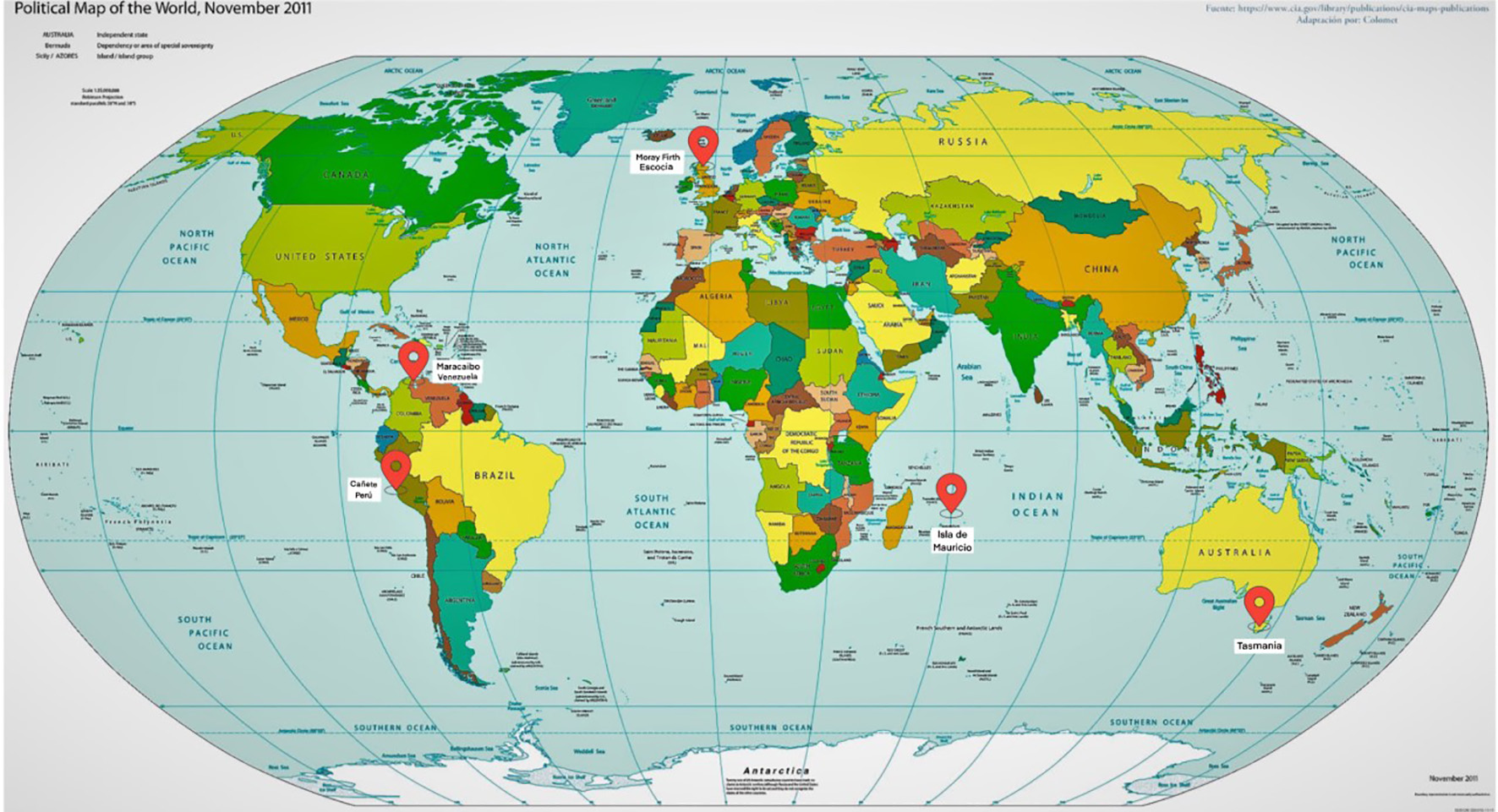
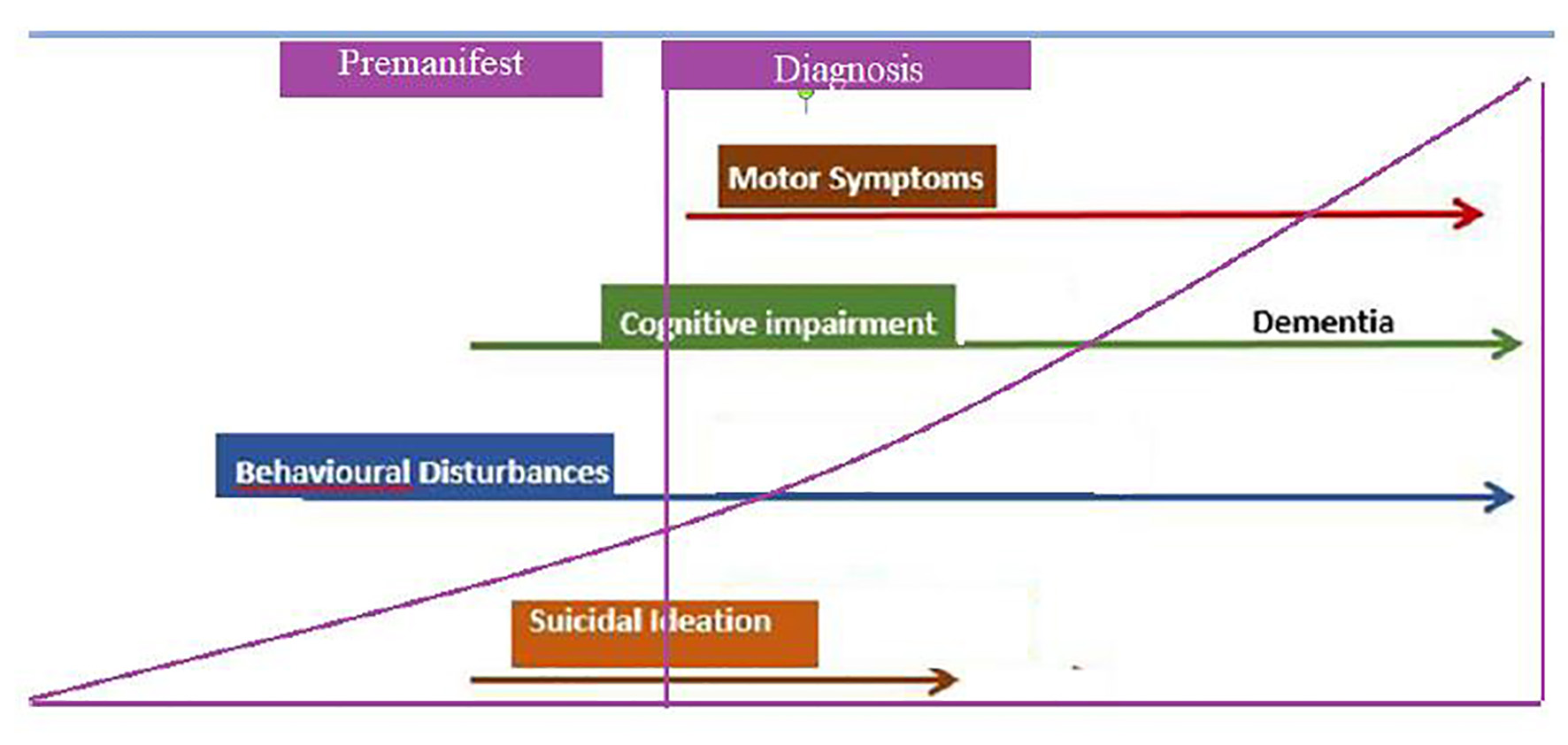
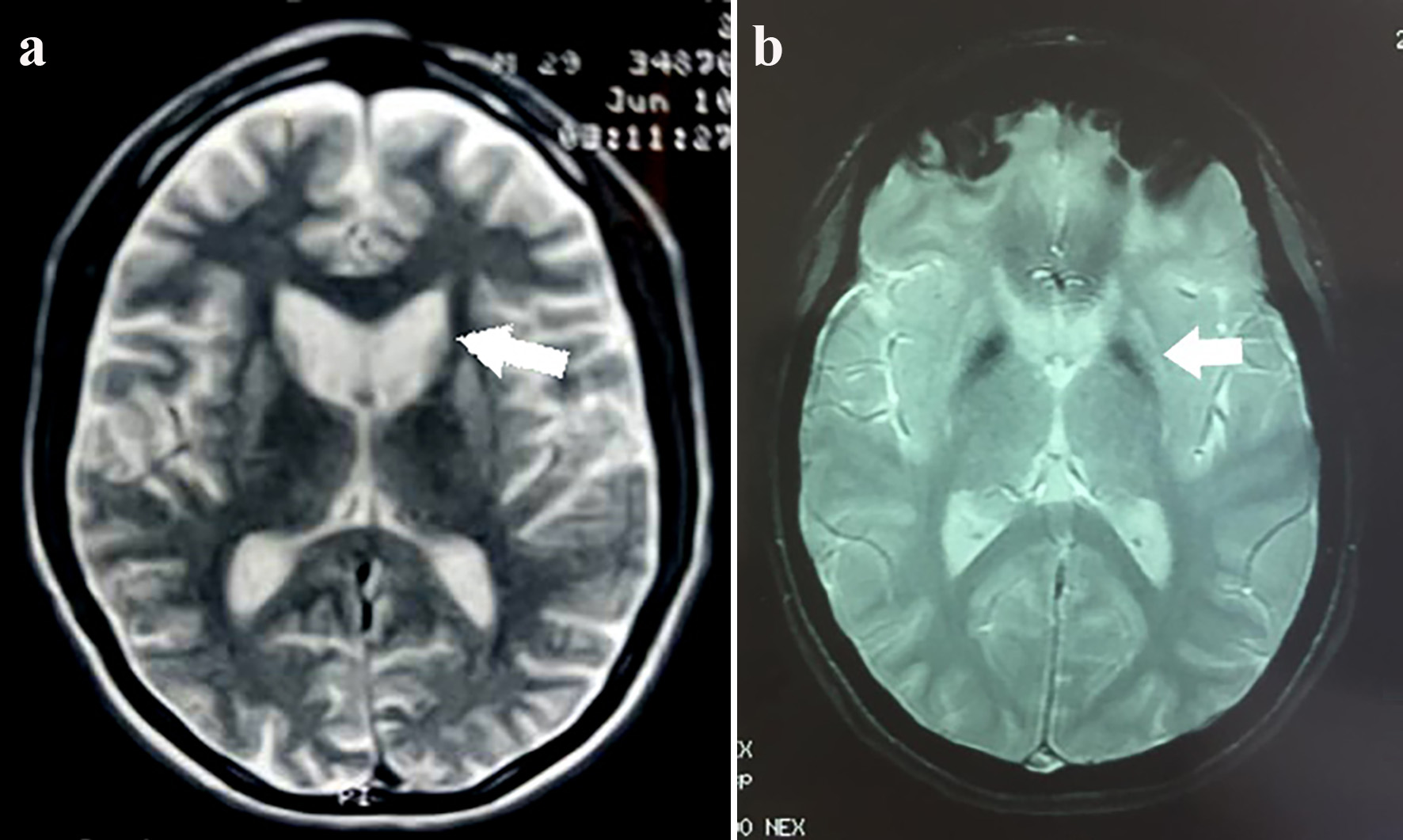
Figure 1. Cluster worldwide populations with the highest prevalence.
| Journal of Neurology Research, ISSN 1923-2845 print, 1923-2853 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Neurol Res and Elmer Press Inc |
| Journal website https://www.neurores.org |
Review
Volume 12, Number 3, October 2022, pages 93-113
Review of Huntington’s Disease: From Basics to Advances in Diagnosis and Treatment
Figures
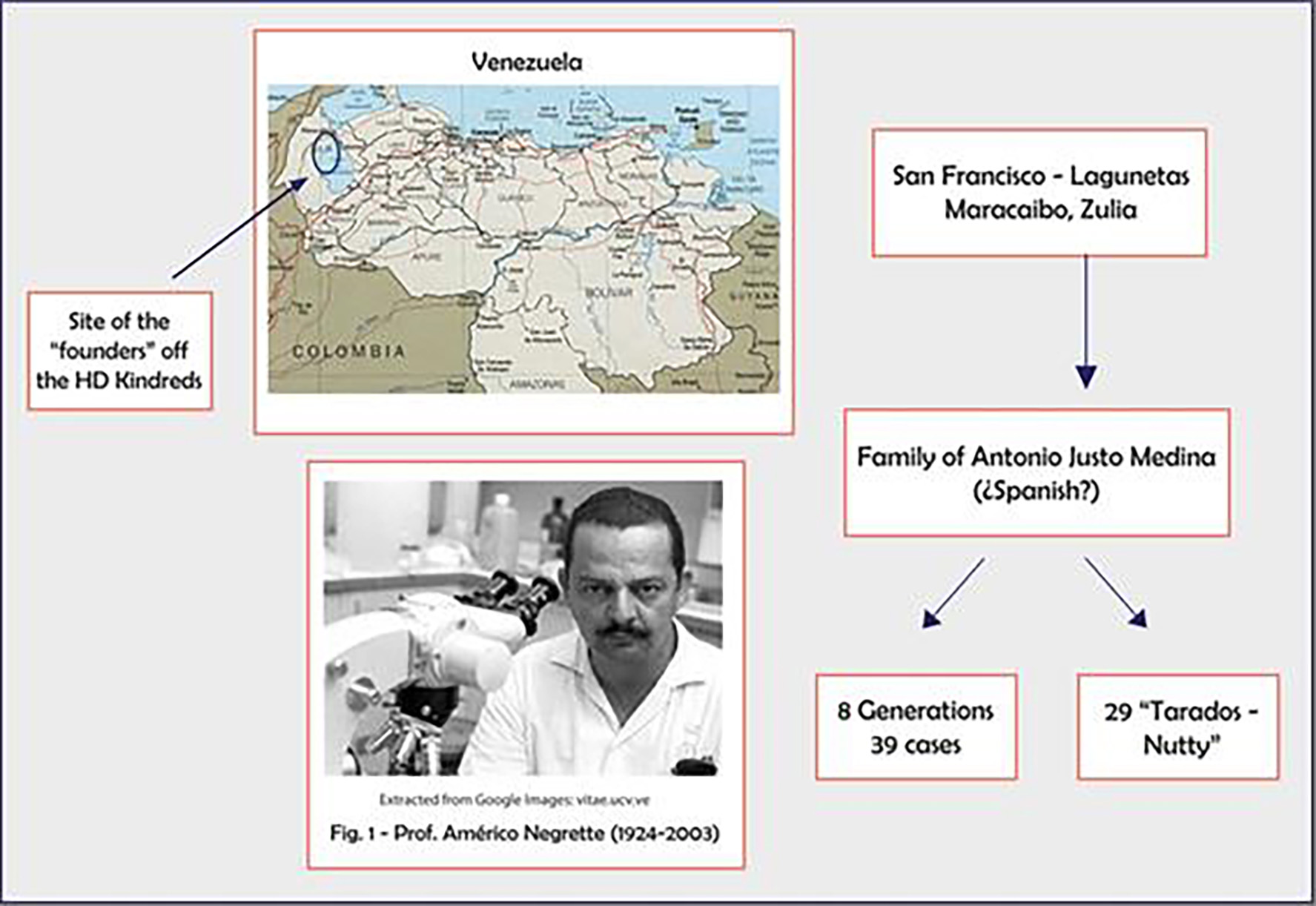
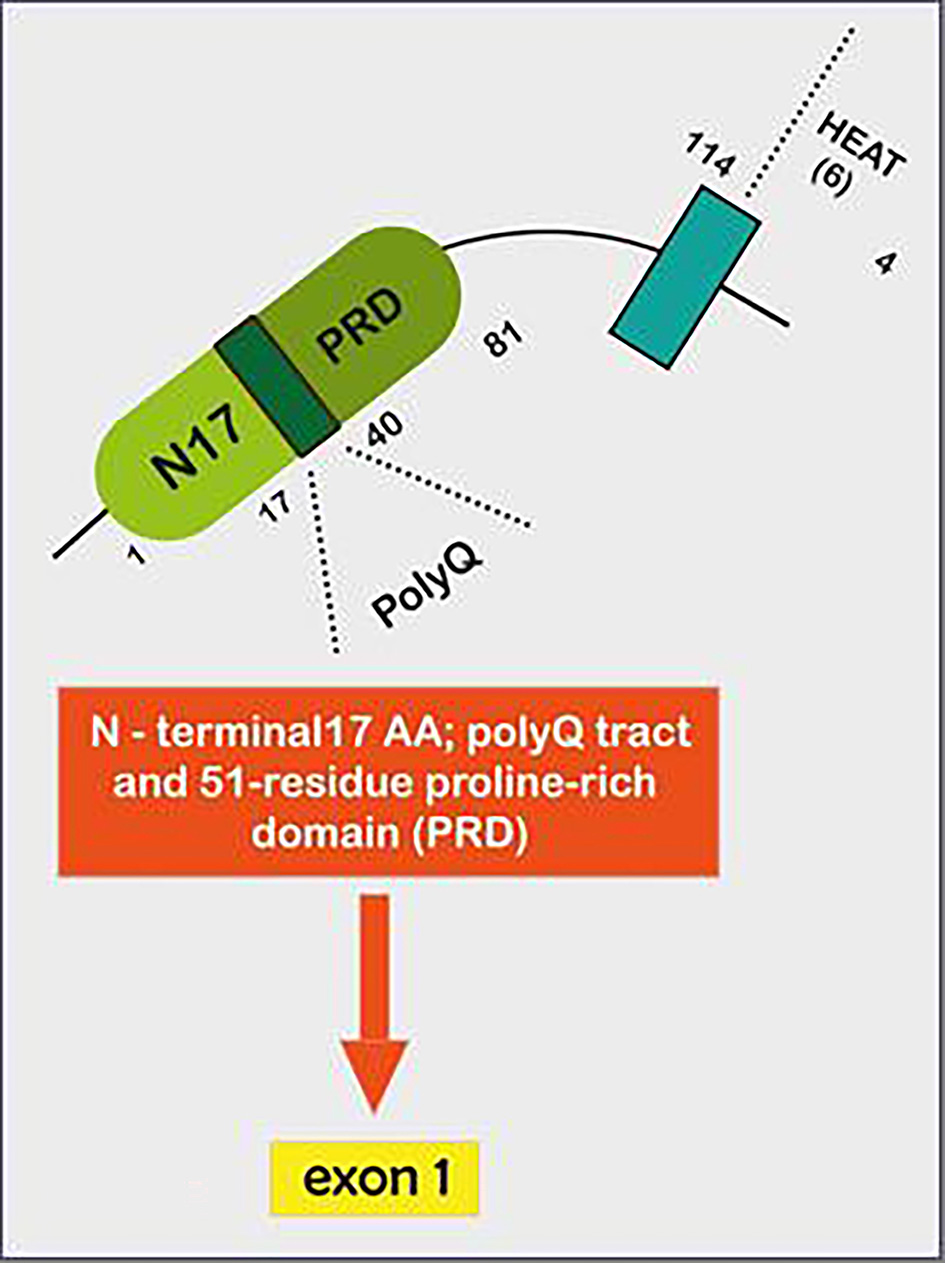
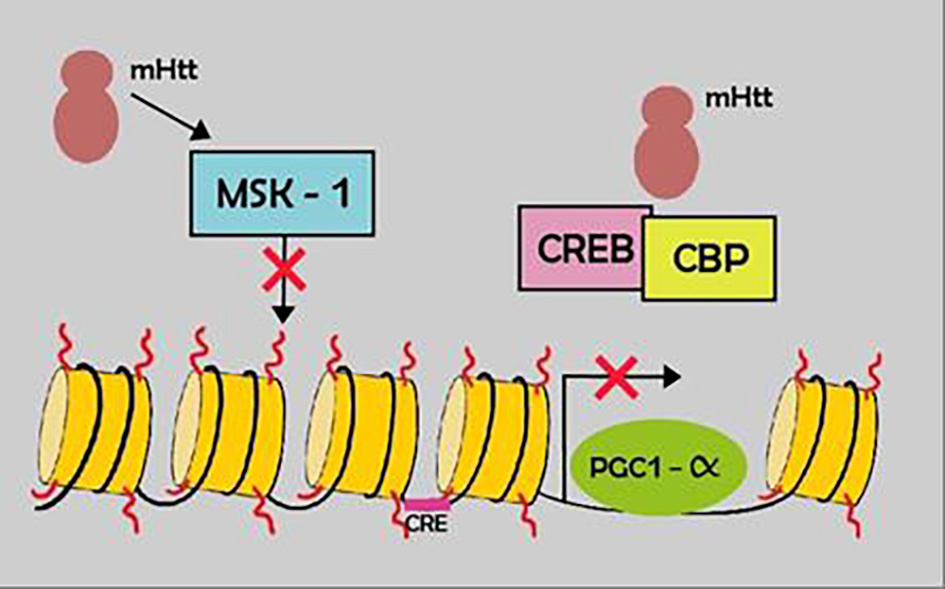
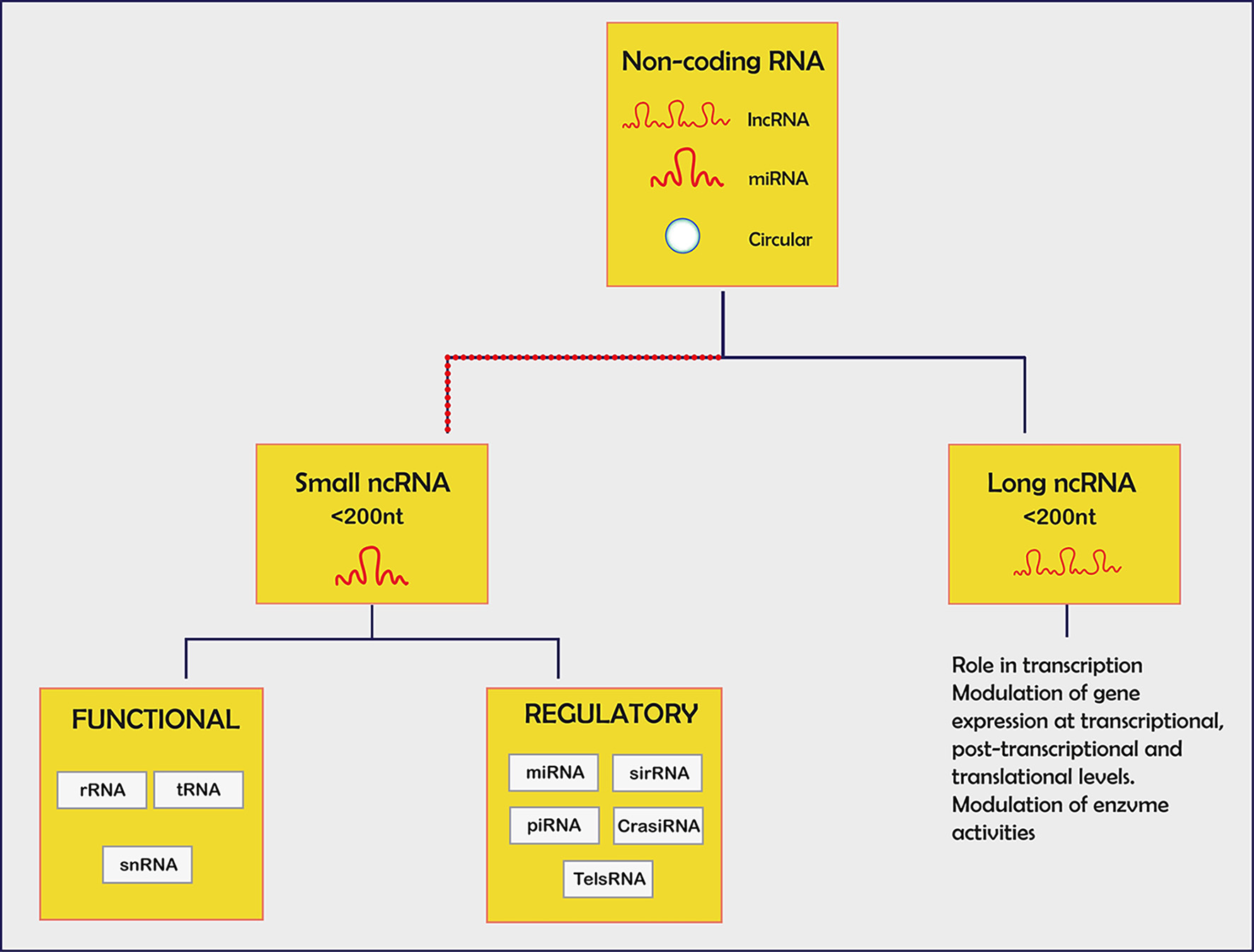
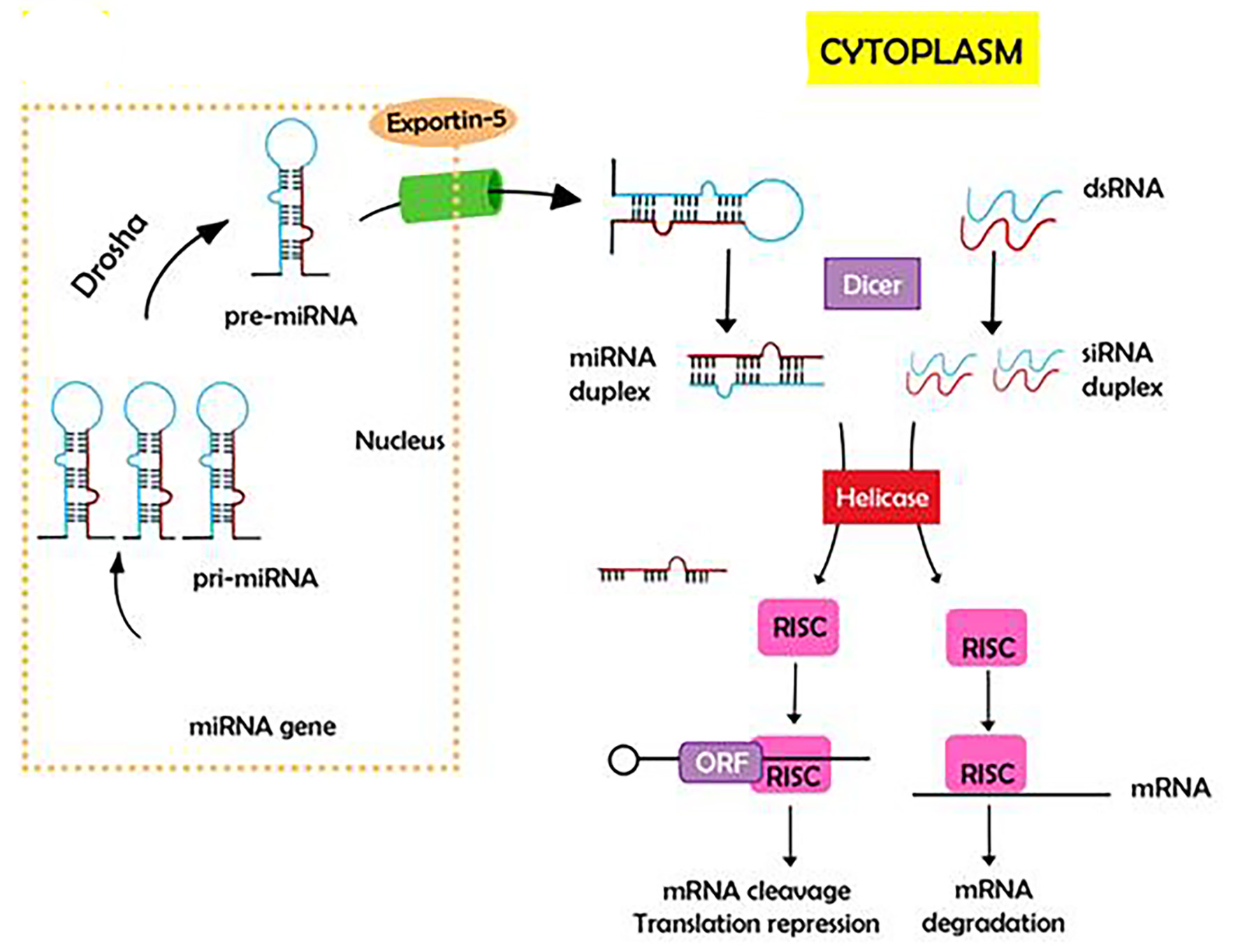
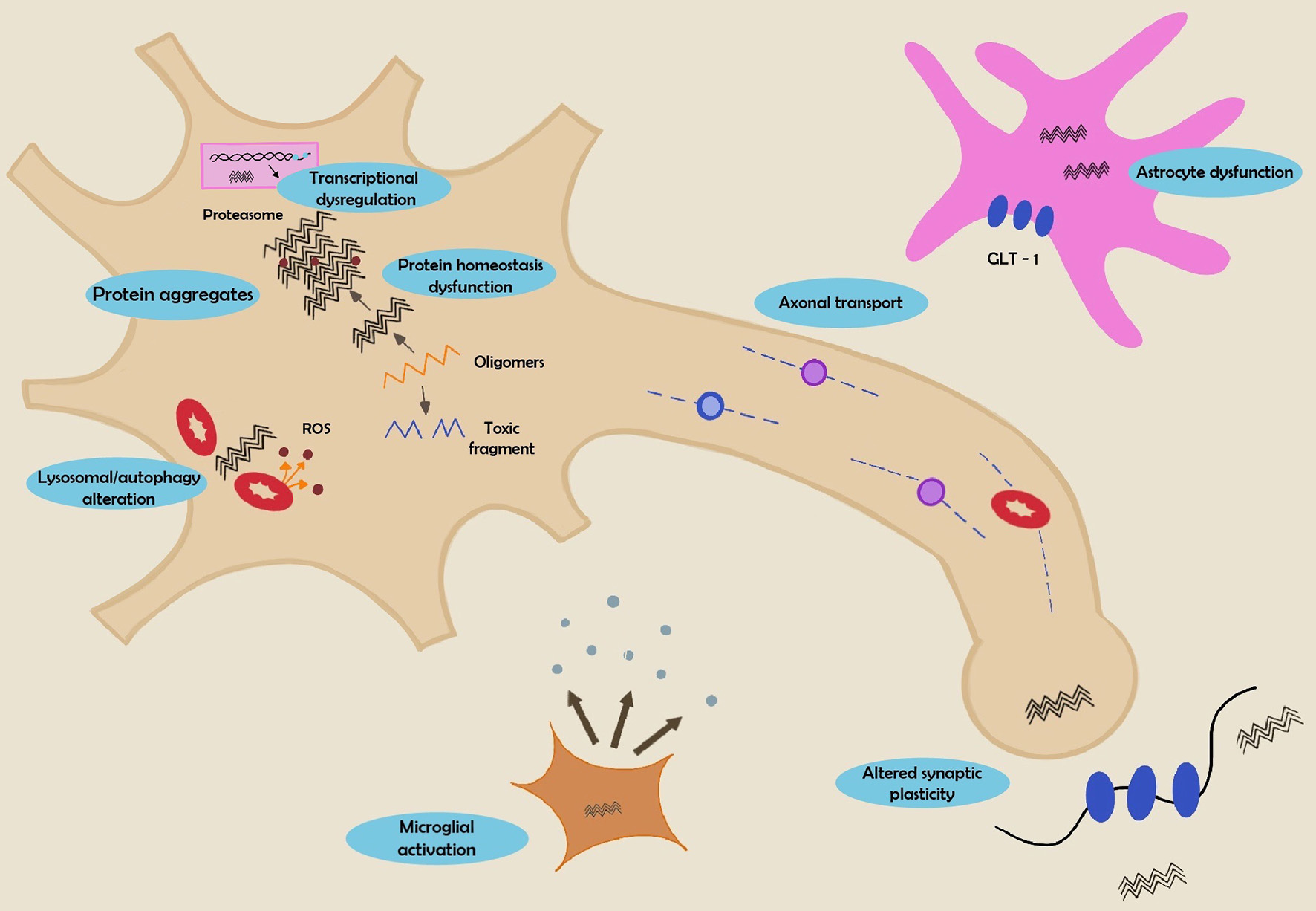
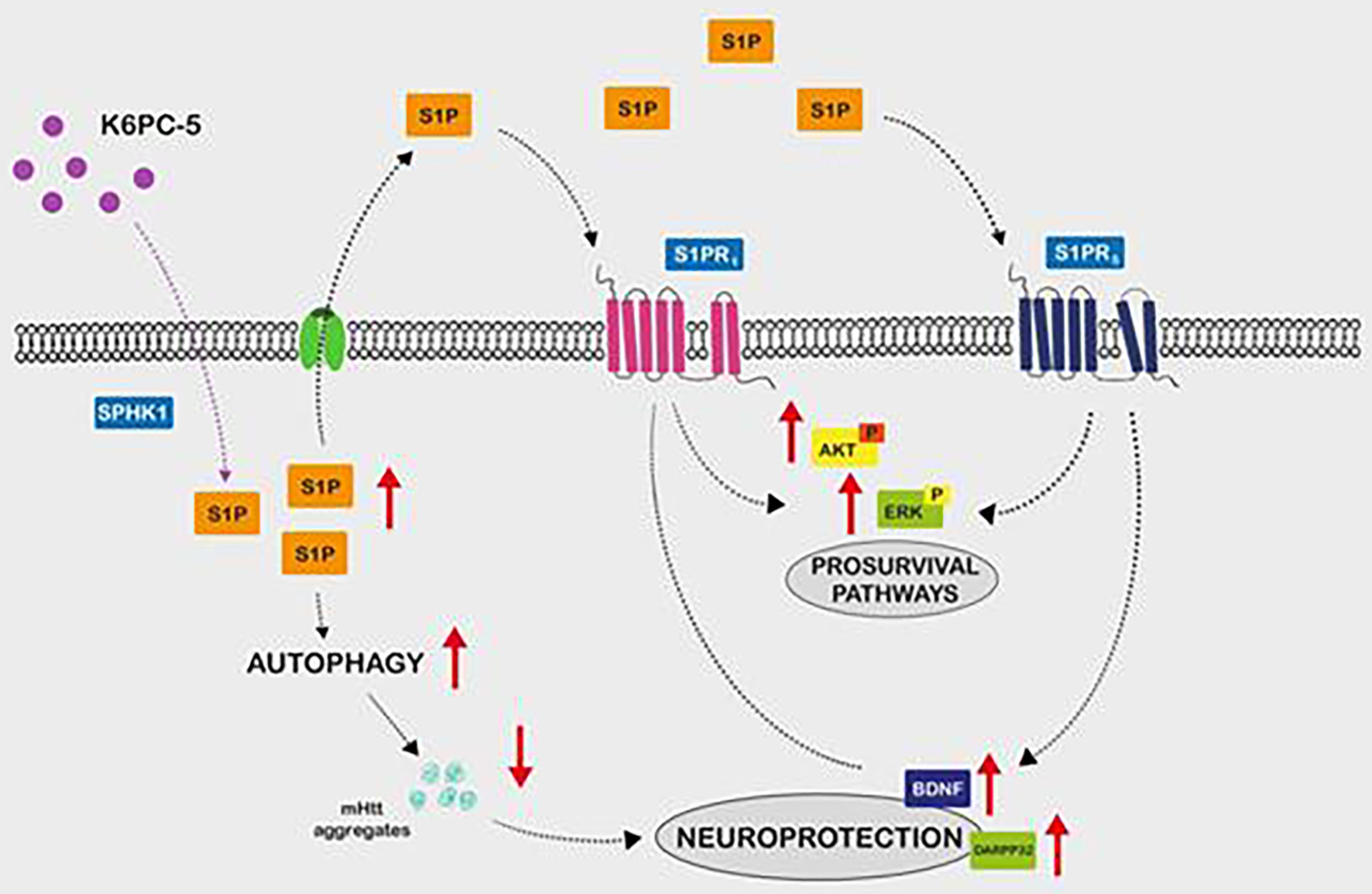
Tables
| SNP ID | Chr | Gene | A1 | A2 | TRACK-HD | Enroll-HD | Meta-analysis | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| β | P | P-FDR | β | P | P-FDR | P-FDR | |||||
| Shown is the genetics association data for the six SNPs genotyped in TRACK-HD and replicated in Enroll-HD. SNPs are ordered by decreasing P-value of their association with the somatic expansion score in the TRACK-HD cohort. Note that in the preliminary analysis using a slightly larger TRACK-HD cohort including four participants with 39 pure CAG repeats (Q1 = 39) and six non-Caucasians, the association between somatic expansion score and rs20579 in LIG1 was P = 0.072 and rs20579 was thus selected for replication in Enroll-HD. Chr: chromosome; A1: minor allele; β: regression coefficient; P: unadjusted P-value; P-FDR: P-value adjusted for multiple testing using the Benjamini-Hochberg false discovery rate correlation; HD: Huntington’s disease; CAG: cytosine-adenine-guanine; HTT: huntingtin. | |||||||||||
| rs3512 | 15 | FAN1 | C | G | 0.06 | 0.003 | 0.034 | 0.05 | 6.7 × 10-5 | 4.0 × 10-4 | 4.8 × 10-6 |
| rs175080 | 14 | MLH3 | A | G | -0.053 | 0.034 | 0.034 | -0.029 | 0.013 | 0.026 | 8.0 × 10-4 |
| rs147804330 | 2 | RP11-481J13,1 | A | G | -0.107 | 0.01 | 0.073 | 0.003 | 0.912 | 0.912 | 0.246 |
| rs1382539 | 5 | MSH3 | A | G | -0.045 | 0.101 | 0.101 | -0.023 | 0.077 | 0.116 | 0.009 |
| rs1799977 | 3 | MLH1 | G | A | -0.032 | 0.093 | 0.314 | -0.034 | 0.009 | 0.026 | 0.004 |
| rs20579 | 19 | MIG1 | A | G | -0.042 | 0.112 | 0.314 | -0.002 | 0.908 | 0.912 | 0.351 |
| Disease | Gene Mt | OMIM | Clinical findings/red flags | Ancestry |
|---|---|---|---|---|
| HDL: Huntington’s disease-like; SCA: spinocerebellar ataxia; DRPLA: dentorubropallidoluisiana; NBIA: neurodegeneration with brain iron accumulation; BHC: Bening hereditary chorea; CA: chorea acanthocytosis; JPH-3: junctophilin-3; TBP: tata box-binding protein; ATXN2: ataxin-2; HRE: hexanucleotide repeat expansion; PRNP: prion protein gene; ATN1: ataxin 1; FTL: ferritin light chain; TITF1: thyroid transcription factor 1; VPS13A: vacuolar protein sorting 13 homolog A. | ||||
| AD | ||||
| HDL2 | JPH-3 | 606438 | Circulating acanthocytes | African |
| SCA 17 | TBP | 607136 | Ataxia chorea | Europe |
| SCA 2 | ATXN2 | 183090 | Nystagmus, impaired saccadic movements and pyramidalism | Brazil |
| C9orf72 | HRE | 614260 | Early psychiatric and behavioral problems pyramidal features | |
| HDL1 | PRPN | 603218 | Chorea prionic disease | African |
| DRPLA | ATN1 | 607462 | Myoclonus, seizures and/or chorea | Japan |
| NBIA3 | FTL | 606159 | Chorea replaces parkinsonism | |
| BHC | TITF1 | 610978 | Childhood disease accompanied by hypothyroidism | |
| AR | ||||
| CA | VPS13A | 200150 | Movement disorders, cognitive decline and behavioral abnormalities | |
| PET | Radioligand | Findings |
|---|---|---|
| PET: positron emission tomography; UHDRS: Unified Huntington Disease Rating Scale; FDG: fluorodeoxyglucose; HD: Huntington’s disease. | ||
| Glucose metabolism | 18F-FDG | Hypometabolism in the caudate and putamen countered by hypermetabolism in thalamic structures at preclinical stages. Striatal and Cortical hypometabolism are associated with motor and cognitive symptoms. Changes may precede neuronal loss. |
| Dopamine D2r | 11C-βCIT; 11C-TBZ presynaptic tracers. 11C-raclopride**/11C FLB457 | **Putamen binding correlations: motor score UHDRS; total functional capacity. Reduction cortical and putaminal binding |
| PDE10A | 18F-MNI-659*** | High expression in the median spinous neurons of the striatum (possible therapeutic target) ***Earliest change in carriers premanifest, even 15 to 20 years prior to clinical manifestations |
| 11C-IMA-107 | ||
| Microglial activation | 11C-PK11195 | Cortical and subcortical areas of significantly raised mean 11C-(R)-PK11195 binding in presymptomatic HD gene carriers compared to controls. Predictive 5-year probability of developing HD. |
| Symptoms | Feature | Target | Drug group | Drug | Doses |
|---|---|---|---|---|---|
| Motor symptoms | Chorea | Reduce dopaminergic neurotransmission | Typical neuroleptics | Haloperidol | 0.5 - 10 mg |
| Atypical neuroleptics | Clozapine | 50 - 150 mg | |||
| Quetiapine | 25 - 200 mg | ||||
| Olanzapine | 2.5 - 20 mg | ||||
| Risperidone | 0.5 - 2 mg | ||||
| Sigma 1 receptor agonist | Pridopidine* | Post-hoc analysis improved TFC in early HD | |||
| Anti-NMDA7 glutamatergic Inhibitor | Amantadine | 100 - 400 mg | |||
| Presynaptic dopamine-depleting agents | Selectively depletes central monoamines | Tetrabenazine | 12.5 - 200 mg | ||
| Deutetrabenazine | 12.5 - 50 mg | ||||
| Valbenazine | 40 - 80 mg | ||||
| Cognitive symptoms | Dementia | Cholinesterase inhibitor | Cholinesterase inhibitor | Rivastigmine | 1.5 - 13.3 mg |
| Inflammation regulation | p38α MAPK inhibition* | Neflamapimod | NCT03980938 | ||
| Anti-glutamatergic | NMDA receptor modulation* | SAGE-718 | NCT03787758 | ||
| Psychiatric symptoms | Depression | SSRIs | Fluoxetine | 20 - 60 mg | |
| Citalopram | 20 - 60 mg | ||||
| Escitalopram | 10 - 20 mg | ||||
| Paroxetine | 20 - 60 mg | ||||
| Sertraline | 50 - 200 mg | ||||
| Tricyclic antidepressants | Tricyclic antidepressants | Nortriptyline | 25 - 150 mg | ||
| Desipramine | 100 - 300 mg | ||||
| Amitriptyline | 75 - 150 mg | ||||
| Atypical antipsychotics | Olanzapine | 2.5 - 10 mg | |||
| Risperidone | 0.5 - 2 mg | ||||
| Aripiprazole | 10 - 30 mg | ||||
| Clozapine | 50 - 150 mg | ||||
| Quetiapine | 25 - 200 mg | ||||
| Obsessive and/or compulsive symptoms | Mood-stabilizing antiepileptic drugs | Antiepileptic drugs | Valproate | 500 - 2,000 mg | |
| Carbamazepine | 200 - 1,600 mg | ||||
| Lamotrigine | 25 - 400 mg | ||||
| Topiramate | 25 - 400 mg | ||||
| Agitation | Benzodiazepines | Clonazepam | 0.5 - 6 mg | ||
| Irritability | Morphinan/class I antiarrhythmic agent | Dextromethorphan/quinidine* | NCT03854019 | ||
| Apathy | Norepinephrine/dopamine reuptake inhibitor | Bupropion | Negative results |