

| Journal of Neurology Research, ISSN 1923-2845 print, 1923-2853 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Neurol Res and Elmer Press Inc |
| Journal website http://www.neurores.org |
Case Report
Volume 4, Number 5-6, December 2014, pages 145-149
Ocular Oscillations and Transient Oscillopsia in Neuromyelitis Optica
Nitin Nemaa, c, Abha Vermaa, Urvija Choudharya, Pramod Sakhib
aDepartment of Ophthalmology, Sri Aurobindo Institute of Medical Sciences and PG Institute, Indore, MP, India
bDepartment of Radiodiagnosis, Index Medical College Hospital & Research Centre, Indore, MP, India
cCorresponding Author: Nitin Nema, Department of Ophthalmology, Sri Aurobindo Institute of Medical Sciences and PG Institute, Indore, MP, India
Manuscript accepted for publication November 11, 2014
Short title: Neuromyelitis Optica
doi: http://dx.doi.org/10.14740/jnr308e
| Abstract | ▴Top |
Neuromyelitis optica (NMO) is a chronic relapsing and disabling disease. It typically causes inflammatory destruction of optic nerve as well as spinal cord that ultimately leads to blindness and paraplegia, respectively. It is more commonly seen in females who are in late thirties and seropositive for anti-NMO-IgG autoantibodies. Ocular movement disorders form an important clinical sign of multiple sclerosis (MS). They are less commonly described in NMO and represent an overt or an occult pathology of brainstem. These ocular movement disturbances generally improve with treatment. However, oscillopsia, with or without nystagmus, is a less reported symptom of NMO that appears early during the course of disease. We are reporting a patient in whom the diagnosis of NMO was made only after the full evolution of the disease, because of the misconception that a closer association exists between MS and optic neuritis and nystagmus. Oscillopsia manifested late during the relapse in this case and disappeared on disease remission although nystagmus persisted throughout the course of disease. We recommend that in patients having nystagmus and optic neuritis a possibility of NMO must be kept in mind.
Keywords: Neuromyelitis optica; Magnetic resonance imaging; T2-weighted imaging; Aquaporin-4; Anti-NMO-IgG antibodies; Intravenous methylprednisolone
| Introduction | ▴Top |
Neuromyelitis optica (NMO) or Devic’s disease is a rare relapsing autoimmune disorder of central nervous system (CNS) that preferentially causes inflammatory destruction of optic nerve and spinal cord [1]. Optic neuritis and myelitis in NMO may occur either simultaneously or sequentially. Brainstem involvement leads to loss of bladder and bowel control, muscle weakness and sensory deficit. Furthermore, it may also cause eye movement abnormalities and oscillopsia [2]. Ocular movements, however, become normal with treatment and so does oscillopsia.
We report a case of recurrent optic neuritis with nystagmus that was misdiagnosed and treated as multiple sclerosis (MS). A definitive diagnosis of NMO could be made only when the disease took a downhill course and permanent physical disability developed in the patient.
| Case Report | ▴Top |
First episode
A 22-year-old female presented with sudden painful loss of vision in her left eye. Her best-corrected visual acuity (BCVA) in right eye (OD) was 6/6 and finger counting (FC) in the left eye (OS). There was relative afferent pupillary defect (RAPD), normal optic disk on funduscopy and defective color vision on Ishihara plates in OS. Gaze-evoked nystagmus was noticed in both eyes (OU). No systemic neurological abnormality was found. Routine hematological investigations were unremarkable. Brain magnetic resonance imaging (MRI) was normal while OS orbital MRI showed hyperintensity in the retrobulbar course of left optic nerve on T2-weighted imaging (Fig. 1). A diagnosis of retrobulbar neuritis (cause MS) OS was made. Intravenous methylprednisolone (IVMP) injections in the dose of 1 g/day were administered for 3 days followed by tapering oral doses of corticosteroids. The patient improved to 6/6 vision with brisk pupillary reaction to light and normal color vision in OS in 4 weeks’ time. However, nystagmus persisted but the amplitude of oscillations was dampened.
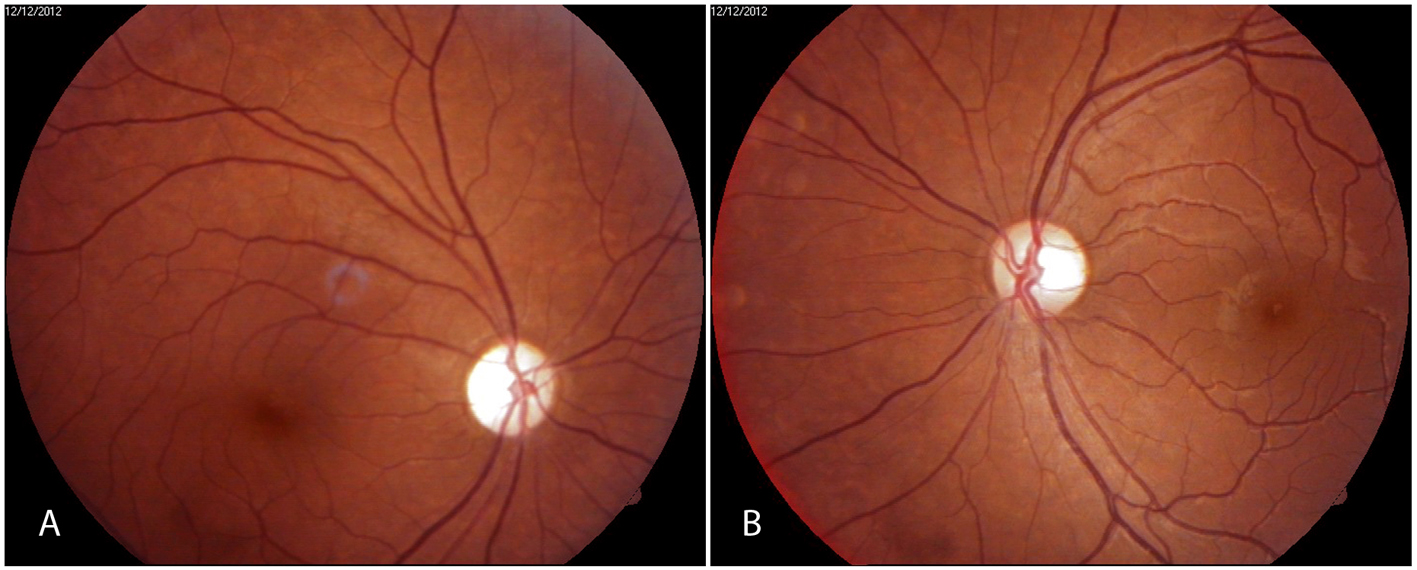 Figure 1. Fundus photographs showing (A) optic atrophy right eye and (B) temporal pallor of optic disk in left eye. Figure 1. Fundus photographs showing (A) optic atrophy right eye and (B) temporal pallor of optic disk in left eye. |
Second episode
The patient remained symptom-free for almost 2 years when she suffered from high grade fever, difficulty in breathing along with weakness in all four limbs, and upper back and chest pain which were found to be due to transverse myelitis affecting cervical-dorsal segments. Occasional bouts of hiccup, on and off nausea and vomiting, and nystagmus were also present. However, other clinical brainstem findings like ptosis, diplopia, hearing loss and facial weakness were not seen. Complete blood count (CBC) was normal except for marginally raised sedimentation rate (30 mm in first hour). Anti-nuclear autoantibody test was non-reactive but thyroid profile revealed hypothyroidism (TSH > 100 mIU/L). Cerebrospinal fluid (CSF) analysis was within normal limits. MRI showed continuous multisegmental, intramedullary longitudinal lesions in the spinal cord extending from C2 to C7 level (Fig. 2), focal area of hyperintensity on T2-weighted imaging in the left occipital periventricular white matter that was inconsistent with MS brain lesion. A diagnosis of MS/NMO was suspected. The patient again received IVMP but had a poor recovery. She developed paraplegia and total sensory loss below T-4 segment.
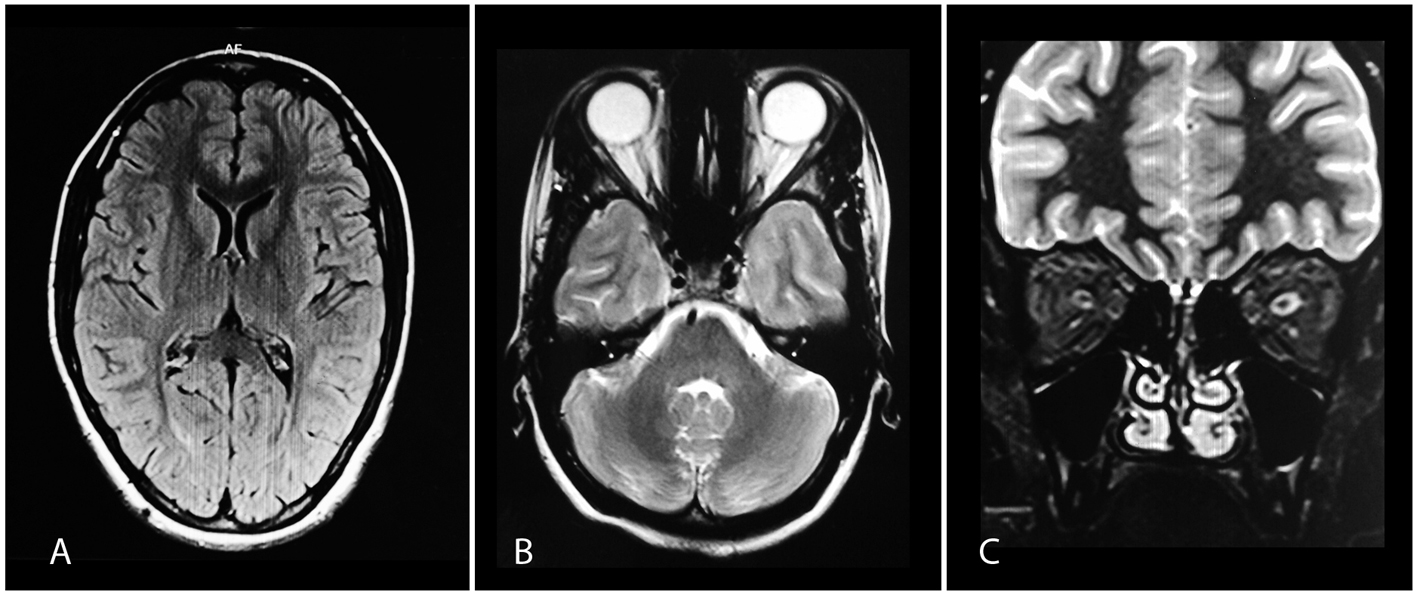 Figure 2. (A) Normal brain MRI on FLAIR. (B, C) Axial and coronal images showing hyperintense intracanalicular parts of left optic nerve on T2-weighted imaging. Figure 2. (A) Normal brain MRI on FLAIR. (B, C) Axial and coronal images showing hyperintense intracanalicular parts of left optic nerve on T2-weighted imaging. |
Third episode
About 3 months after the first relapse, the patient had an episode of loss of vision in OD. MRI spine showed an abnormal hyperintense mildly enhancing intramedullary signal from C7 to D4 vertebral level, and MRI brain showed few small non-enhancing lesions in juxtacortical part of frontal and parietal lobes and T2-weighted hyperintensity in subependymal wall of right occipital trigone which were atypical for MS. Abnormal hypersignal intensity in intraorbital and intracanalicular parts of right optic nerve were noted on MRI orbit which showed homogeneous enhancement on post contrast study favoring the diagnosis of optic neuritis and marginal left optic nerve atrophy. NMO-IgG antibody was found to be positive that ultimately established the diagnosis of NMO. Despite treatment with IVMP, the patient had a poor recovery in OD. Oral azathioprine 150 mg/day was started along with low dose steroids.
Fourth episode
The patient developed another attack of optic neuritis in OS about 3 years from presentation. The vision in OD was hand movement (HM) and FC in OS. Exotropia OD and gaze-evoked nystagmus on lateral gaze OU were present. There was APD with ill-sustained pupillary reactions OU. Fundus examination showed generalized optic disk pallor in OD and temporal disk pallor in OS. CBC and CSF analysis were normal. Blood test for thyroid antibodies was positive for antithyroid peroxidase antibodies. However, anti-dsDNA antibody test was negative. Neuroimaging study with contrast showed non-enhancing white matter lesions of frontal and parietal lobes of brain and enhancing plaque in canalicular part of right optic nerve. The T2-weighted imaging revealed atrophy of upper and mid dorsal cord (from C7 to D6 level) on MRI spine (Fig. 3A) and hyperintensity signals in intracanalicular portion of right optic nerve with bilaterally thinned and atrophied optic nerves (Fig. 3B). The patient was given IVMP 1 g/day for 5 days followed by tapering doses of oral prednisolone and tablet azathioprine. Her OS vision recovered to 6/9 within 10 days. It is at this time she started complaining of oscillopsia, a visual sensation of swaying or movement of environment. As nystagmus got dampened with treatment in the next 3 weeks oscillopsia disappeared.
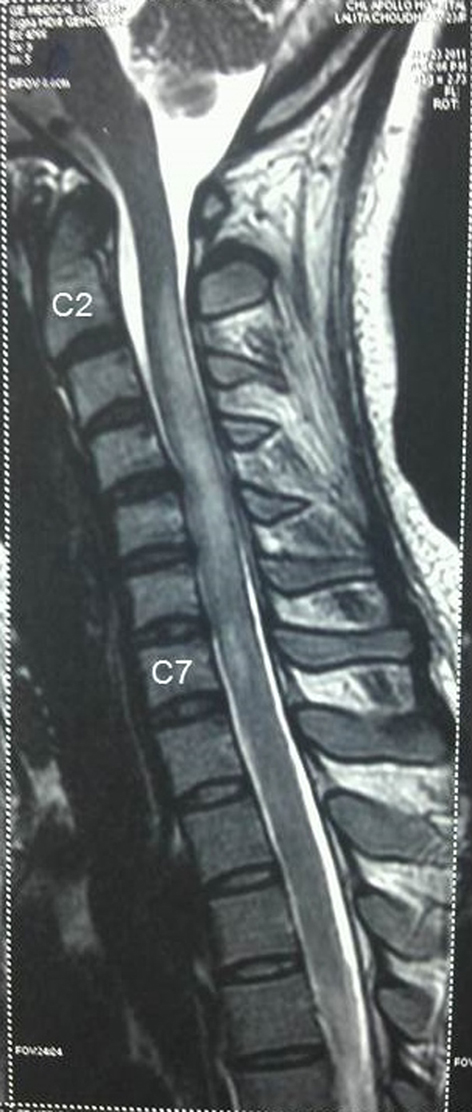 Figure 3. MRI spine showing longitudinally extensive increased signal intensity of spinal cord from C2 to C7 on T2-weighted imaging with non-involvement of brainstem. Figure 3. MRI spine showing longitudinally extensive increased signal intensity of spinal cord from C2 to C7 on T2-weighted imaging with non-involvement of brainstem. |
The last follow-up visit in May 2014 showed BCVA of HM in OD and 6/9 in OS. There was RAPD in OD with normal color vision in OS. Dilated fundus examination showed absence of vitreous cells, normal retinal vasculature with optic disks showing atrophy in OD and temporal pallor in OS (Fig. 4). There was generalized depression of light sensitivity in OS on automated perimetry. Optical coherence tomography revealed more pronounced generalized loss of peripapillary retinal nerve fiber layer in OD as compared to OS. Systemically the patient had paraplegia along with loss of bladder and bowel control, and she needed support for ambulation. She was on daily oral azathioprine (150 mg), prednisolone (40 mg), thyroxine (100 μg) and supportive therapy. She was also receiving active physiotherapy to prevent bed sores and muscle contractures.
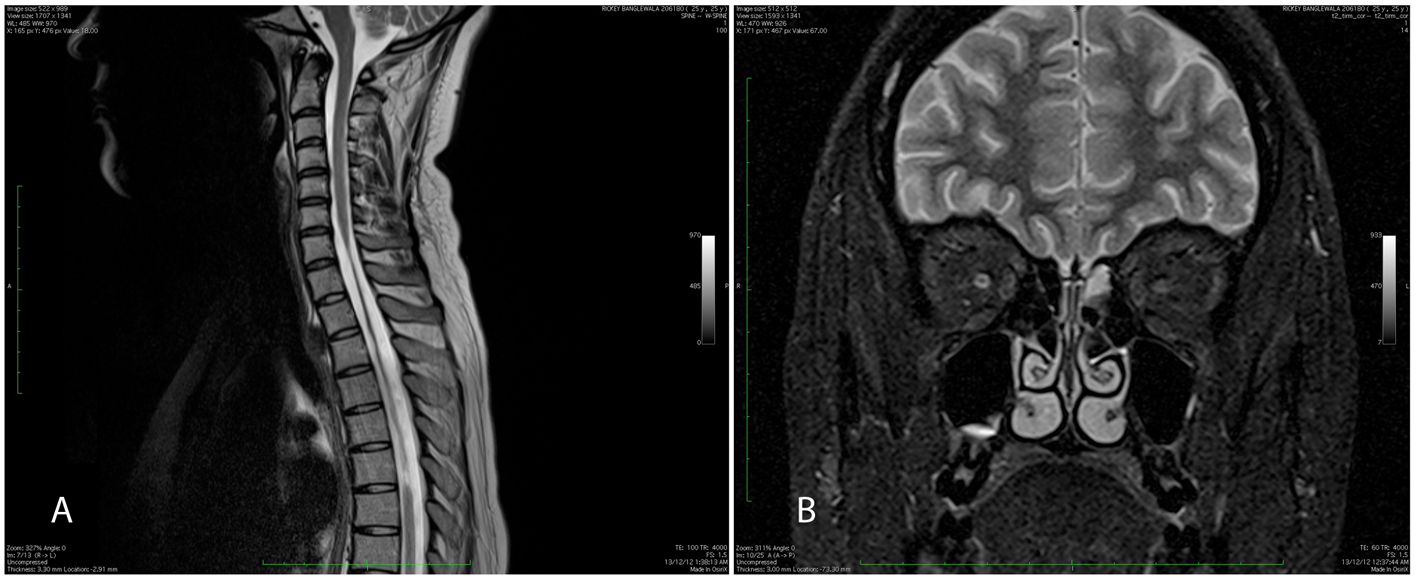 Figure 4. (A) MRI spine showing cord atrophy from C7 to D6 on T2-weighted imaging. (B) Hyperintensity in intracanalicular portion of right optic nerve with bilateral thinning of optic nerves. Figure 4. (A) MRI spine showing cord atrophy from C7 to D6 on T2-weighted imaging. (B) Hyperintensity in intracanalicular portion of right optic nerve with bilateral thinning of optic nerves. |
| Discussion | ▴Top |
NMO is caused by NMO-IgG (aquaporin-4) antibodies. Anti-NMO-IgG (or anti-aquaporin-4, anti-AQP4) antibody is considered as a specific marker for NMO. The antibodies target the aquaporin-4, a protein which acts as main water transport channel of CNS across the astrocyte cell membrane [3]. Aquaporin-4 is most strongly expressed in the optic nerves, spinal cord, hypothalamus and brainstem; these areas are predominantly affected by NMO [4].
NMO causes necrotizing demyelination of optic nerve and spinal cord resulting in rapid and severe visual and physical deficit. The typical relapsing-remitting (polyphasic) type of disease is more common and more fulminant than monophasic. Each relapse results in incomplete recovery and permanent disability. Within 5 years of disease onset, more than 50% patients with relapsing type develop blindness in one or both eyes and need ambulatory help.
Wingerchuk et al’s revised diagnostic criteria remain the cornerstone for the diagnosis of NMO [5]. It is essential to have two absolute and two out of three supportive criteria for making a diagnosis NMO. The absolute criteria include optic neuritis and myelitis. The supportive criteria are presence of contiguous spinal cord MRI signal abnormality extending over three or more vertebral segments, brain MRI not meeting diagnostic criteria for MS at disease onset, and NMO-IgG seropositive status.
Our case met the diagnostic criteria for NMO. The patient had optic neuritis and transverse myelitis, and the spinal cord was involved with lesion extending longitudinally over three contiguous vertebral segments (from C7 to D4 level). Moreover, she was seropositive for NMO-IgG and her brain lesions on MRI were not characteristic of that seen in MS.
NMO is relatively more common in non-Caucasians, Asians and Indians [6, 7]. The median age for onset of the disease is late thirties. It is more represented in females as compared to males.
Some NMO-IgG seropositive patients may have other coexisting autoimmune diseases, most common being systemic lupus erythematosus, Sjogren’s syndrome and rheumatoid arthritis. Less commonly autoimmune thyroid disease may also co-occur [8, 9]. Systemic autoimmune diseases are more strongly associated with polyphasic than monophasic NMO [10, 11]. Their presence is a predictor of poor treatment outcome.
The present case was positive for antithyroid peroxidase antibodies. The course of NMO was aggressive, and the patient developed blindness in one eye and severe disability within 2 years of disease onset. The first episode of optic neuritis was successfully managed with complete visual recovery; however, the subsequent episodes (of optic neuritis and transverse myelitis) resulted in permanent disability due to delayed diagnosis and initiation of immunosuppressive therapy.
About 15% of NMO patients have neurological symptoms indicating disease beyond the optic nerve and spinal cord [7, 11]. In 10% of cases hypothalamus and brainstem are affected that may cause abnormal ocular movements [12]. Ocular movements generally normalize with treatment [2]. Most of these patients with brainstem and hypothalamic symptoms have normal brain MRI and positive NMO-IgG antibodies [2, 5]. Brain MRI fails to detect any pathology because signal of the lesion gradually disappears over time in the same manner as that observed in spinal cord lesions of NMO [13, 14]. Kim et al reported that occult or subtle brain damage in NMO is easily missed on conventional MRI, and to detect such cranial lesion more advanced imaging techniques are required [14]. In a recent study, Kister et al found brain lesions in all 10 patients of NMO they studied using ultrahigh-field MRI [15]. Non-specific brain lesions were detected on cranial MRI in the present case as well.
Like ocular movement disorders, oscillopsia is also a brainstem sign of NMO [2]. Kim et al, however, reported cortical oscillopsia without nystagmus as an initial manifestation of the disease [16]. In contrary, oscillopsia in the present case developed during the relapse of the disease and was a late manifestation. It disappeared with treatment and improvement in the patient’s condition. However, nystagmus that appeared at the disease onset got dampened with treatment but persisted throughout.
Neuromyelitis Optica Study Group recommended prompt initiation of immunosuppressive treatment on confirmation of diagnosis of NMO with azathioprine or rituximab, either alone or in combination with systemic corticosteroids [1]. Watanabe et al reported relapses resistant to corticosteroid therapy mainly in NMO-IgG-seropositive patients [17]. Our patient was on azathioprine and corticosteroids but had to stop corticosteroid for nearly a month due to severe gastritis, and, therefore, developed the third relapse.
The present case highlights some of the commonly made mistakes by clinicians. Cranial MRI along with the imaging of spinal cord must be ordered in any young female patient who presents with optic neuritis and nystagmus with probable demyelinating etiology. Even with disease remission, the patient should be informed about the probability of recurrences. Miller et al recommended periodic surveillance with brain and spinal MRI scanning in these patients [18]. Therefore, such cases with ocular movement disorders must not be written off as MS and should be properly investigated for NMO.
Conflict of Interest
Nil.
Source of Funding
Nil.
| References | ▴Top |
- Trebst C, Jarius S, Berthele A, Paul F, Schippling S, Wildemann B, Borisow N, et al. Update on the diagnosis and treatment of neuromyelitis optica: recommendations of the Neuromyelitis Optica Study Group (NEMOS). J Neurol. 2014;261(1):1-16.
doi pubmed - Hage R, Jr., Merle H, Jeannin S, Cabre P. Ocular oscillations in the neuromyelitis optica spectrum. J Neuroophthalmol. 2011;31(3):255-259.
doi pubmed - Wingerchuk DM. Neuromyelitis optica. Int MS J. 2006;13(2):42-50.
pubmed - Pittock SJ, Weinshenker BG, Lucchinetti CF, Wingerchuk DM, Corboy JR, Lennon VA. Neuromyelitis optica brain lesions localized at sites of high aquaporin 4 expression. Arch Neurol. 2006;63(7):964-968.
doi pubmed - Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology. 2006;66(10):1485-1489.
doi pubmed - Kira J. Multiple sclerosis in the Japanese population. Lancet Neurol. 2003;2(2):117-127.
doi - Sellner J, Boggild M, Clanet M, Hintzen RQ, Illes Z, Montalban X, Du Pasquier RA, et al. EFNS guidelines on diagnosis and management of neuromyelitis optica. Eur J Neurol. 2010;17(8):1019-1032.
doi pubmed - Pittock SJ, Lennon VA, de Seze J, Vermersch P, Homburger HA, Wingerchuk DM, Lucchinetti CF, et al. Neuromyelitis optica and non organ-specific autoimmunity. Arch Neurol. 2008;65(1):78-83.
doi pubmed - Kim W, Kim SH, Kim HJ. New insights into neuromyelitis optica. J Clin Neurol. 2011;7(3):115-127.
doi pubmed - Wingerchuk DM, Weinshenker BG. Neuromyelitis optica: clinical predictors of a relapsing course and survival. Neurology. 2003;60(5):848-853.
doi - Wingerchuk DM, Hogancamp WF, O’Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic’s syndrome). Neurology. 1999;53:1107-1114.
doi - Pittock SJ, Lennon VA, Krecke K, Wingerchuk DM, Lucchinetti CF, Weinshenker BG. Brain abnormalities in neuromyelitis optica. Arch Neurol. 2006;63(3):390-396.
doi pubmed - Cassinotto C, Deramond H, Olindo S, Aveillan M, Smadja D, Cabre P. MRI of the spinal cord in neuromyelitis optica and recurrent longitudinal extensive myelitis. J Neuroradiol. 2009;36(4):199-205.
doi pubmed - Kim W, Kim SH, Huh SY, Kim HJ. Brain abnormalities in neuromyelitis optica spectrum disorder. Mult Scler Int. 2012;2012:735486.
- Kister I, Herbert J, Zhou Y, Ge Y. Ultrahigh-Field MR (7 T) Imaging of Brain Lesions in Neuromyelitis Optica. Mult Scler Int. 2013;2013:398259.
- Kim SM, Kim JS, Heo YE, Yang HR, Park KS. Cortical oscillopsia without nystagmus, an isolated symptom of neuromyelitis optica spectrum disorder with anti-aquaporin 4 antibody. Mult Scler. 2012;18(2):244-247.
doi pubmed - Watanabe S, Nakashima I, Misu T, Miyazawa I, Shiga Y, Fujihara K, Itoyama Y. Therapeutic efficacy of plasma exchange in NMO-IgG-positive patients with neuromyelitis optica. Mult Scler. 2007;13(1):128-132.
doi pubmed - Miller DH, Weinshenker BG, Filippi M, Banwell BL, Cohen JA, Freedman MS, Galetta SL, et al. Differential diagnosis of suspected multiple sclerosis: a consensus approach. Mult Scler. 2008;14(9):1157-1174.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Neurology Research is published by Elmer Press Inc.