

| Journal of Neurology Research, ISSN 1923-2845 print, 1923-2853 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Neurol Res and Elmer Press Inc |
| Journal website http://www.neurores.org |
Case Report
Volume 7, Number 4-5, October 2017, pages 94-96
DiGeorge Syndrome Presenting With Seizures: A Case Report
Samaneh Noroozi Asla, Hoda Khatib Masjedib, c, Ali Rajabpour Sanatib
aFaculty of Medicine, Mashhad University of Medical Sciences, Mashhad, Iran
bFaculty of Medicine, Birjand University of Medical Sciences, Birjand, Iran
cCorresponding Author: Hoda Khatib Masjedi, 2nd Floor, Apartment #68, Guya 4, Malekabad Blv., Mashhad, Razavi Khorasan, Iran
Manuscript submitted November 4, 2016, accepted December 5, 2016
Short title: DiGeorge Syndrome With Seizures
doi: https://doi.org/10.14740/jnr410e
| Abstract | ▴Top |
DiGeorge syndrome was described for the first time in 1968 as a defect affecting structures derived from the third and fourth embryonic pharyngeal arches along with absent parathyroid glands. According to the low incidence of this disease as well as a wide spectrum of symptoms, it is essential to report cases with less prevalent features. In this case report, a child was introduced with a diagnosis of DiGeorge syndrome presenting with seizures. The patient was a 27-day-old baby girl due to seizures admitted to Hospital Imam Reza (AS), Mashhad, Iran. Hypocalcemia was observed in early clinical trials requested. The patient underwent echocardiography according to holosystolic murmur grade 3/6 auscultation, which showed a patent ductus arteriosus (PDA), tetralogy of Fallot (TOF), ventricular septal defect (VSD), atrial septal defect (ASD), and pulmonary atresia (PA). No thymus was found on chest X-ray, and evidence of previous conflicts was observed in the heart. Finally, fluorescent in situ hybridization (FISH) was performed to check out Tuple gene deletion on chromosome 22q11.2, and the diagnosis was confirmed for DiGeorge syndrome. Although the incidences of neurological symptoms associated with hypocalcemia suggest a wide range of diseases as a differential diagnosis, pediatrics should consider the heart disorders for DiGeorge syndrome through clinical examinations and imaging, if necessary.
Keywords: DiGeorge syndrome; Gene Tuple deletion; Neurological symptoms; Seizures
| Introduction | ▴Top |
DiGeorge syndrome is one of the most common syndromes associated with chromosomal q11.222 deletion, and its incidence is approximately 1 in 4,000 live births [1]. DiGeorge syndrome is a collection of heart abnormalities, such as tetralogy of Fallot (TOF) and truncus arteries (TA) along with hypoparathyroidism, and thymic hypoplasia (and secondary immunodeficiency) [2]. However, other syndromes can be seen, such as CHARGE syndrome, conotruncal anomaly face syndromes, and CATCH (velocardiofacial shprintzen) with chromosome q11.222 deletion [1-3]. DiGeorge syndrome was described for the first time in 1968 as a defect affecting structures derived from the third and fourth embryonic pharyngeal arches along with absent or small parathyroid glands [4]. Many of the patients involved in the neonatal period die due to heart abnormalities and infections. However, the diagnosis does not take place properly most of the time due to the low incidence of this disease as well as the broad and varied spectrum of symptoms. In this case report, we introduced a child with a diagnosis of DiGeorge syndrome presenting with seizures.
| Case Report | ▴Top |
Present illness and medical history
The case was a 27-day-old girl, who was admitted to Imam Reza (AS) Hospital, Mashhad, Iran due to seizures. Seizure appeared as repeated myoclonic attacks along with 10 s upward gaze of eyes, which had been started 5 days ago. Birth weight was 3,200 g with a head circumference of 35 cm at birth. The patient was born by cesarean delivery in her 38th week with normal Apgar score at birth. Parents had no family history of diseases, and mother gravidity was four, with four alive children.
Examinations
Patient weight was 3,900 g. The patient had central cyanosis at admission and a holo systolic murmur grade 3/6 was auscultated. The other systems examination was normal. The baby was transferred to the neonatal intensive care unit due to a low-grade fever and seizure and underwent a sepsis workup.
Experiments
Blood cell count was normal. Venous blood gases (VBG: pH: 7.32, 3HCo: 21.5, PCO2: 40, PO2: 33.6) was compatible with the normal VBG. The patient underwent lumbar puncture and cerebrospinal fluid (CSF) test results represented the white blood cells: 0 - 1 mm3, red blood cells: 0 - 1 mm3, lactate dehydrogenase (LDH): 90 U/L, protein: 68 mg/dL, and glucose: 61 mg/dL. Blood, urine, and CSF cultures were all negative. The erythrocyte sedimentation rate (ESR) was 28 mm/h, and C-reactive protein (CRP) was single positive. Other results were calcium blood level: 6.7 mg/dL, phosphorus: 9.9 mg/dL, magnesium: 1.8 mg/dL, alkaline phosphatase: 797 IU/mL, parathyroid hormone (PTH): 14 pg/mL, 25(OH) D: 15 ng/mL, and albumin: 3.8 g/dL.
Imaging procedures
Brain magnetic resonance imaging (MRI) and electroencephalography (EEG) results were both normal, which were done because of the seizure. Also, an echocardiography was performed, and the results were reported by patent ductus arteriosus, TOF, ventricular septal defect, atrial septal defect and pulmonary atresia. The following evidences were found on chest X-ray (Fig. 1): lack of thymic visibility, narrow mediastinum, heart enlargement with right ventricle (RV) configuration, narrow heart pedicle, and a lung field hypoperfusion.
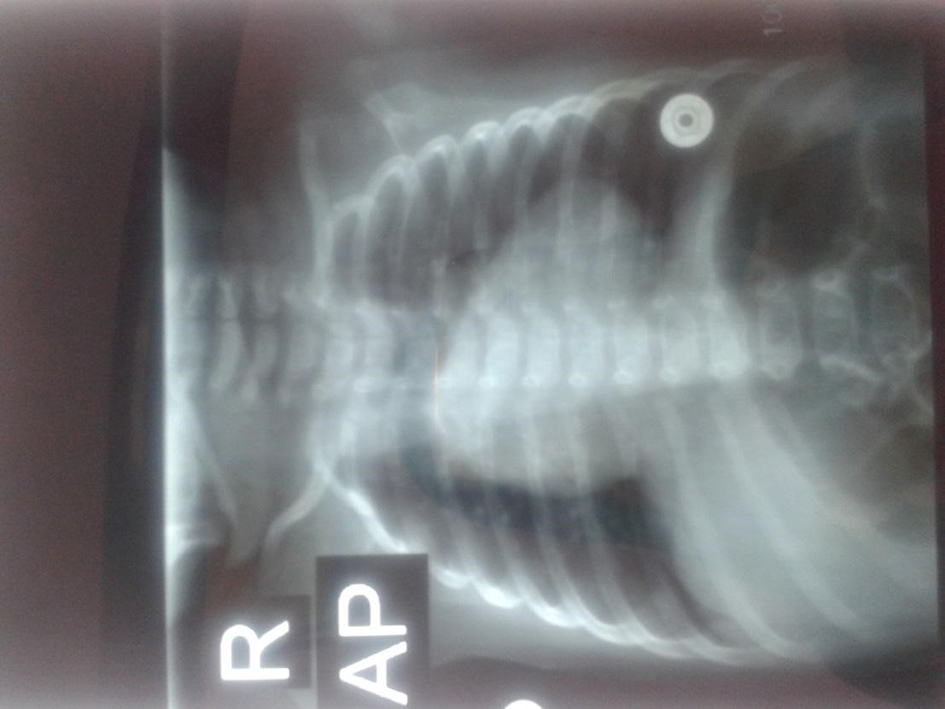 Click for large image | Figure 1. Chest X-ray for the patient suspected to DiGeorge syndrome. |
Follow-up
DiGeorge syndrome was assumed according to the results of tests, echocardiography, and clinical suspicion. For a definitive diagnosis, peripheral blood samples were prepared to assess Tuple (Hira) gene region deletion on chromosome 22q11.2 using fluorescent in situ hybridization (FISH) technique. The chromosomal study confirmed the diagnosis of DiGeorge syndrome.
| Discussion | ▴Top |
The most important preliminary finding in the child is hypocalcemia. Hypocalcemia can be due to neonatal or maternal causes. Maternal causes include diabetes mellitus, maternal vitamin D deficiency, maternal hyperparathyroidism, use of antiepileptic drugs by mother, excessive intake of magnesium by mother and preeclampsia. Neonatal causes include low birth weight, prematurity, IUGR, asphyxia, sepsis, hyperbilirubinemia and its treatment (phototherapy or exchange transfusion), hypomagnesemia and hypermagnesemia, hypoparathyroidism, vitamin D deficiency and osteopetrosis [5]. Neonatal hypoparathyroidism is more likely than others due to lower PTH levels in the child. The most important congenital causes of neonatal hypoparathyroidism are aplasia of parathyroid glands, which is common in DiGeorge syndrome [5]. This syndrome is detected at a rate of 1/4,000 newborns, and in 90% of cases is caused by chromosome 22q11.2 deletion. About 25% of children inherit this disorder from their parents. Neonatal hypocalcemia can be seen in 60% of patients involved, but often temporary; however, it can be associated with recurrent courses. Other associated causes include Barakat syndrome, Kenny-Caffey syndrome and lymphedema, so patients must be carefully evaluated regarding these clinical examinations as well as paraclinical investigations. In patient examined in the present study, concerning associated cardiac abnormalities, especially different conotruncal, the most likely diagnosis was for DiGeorge syndrome which was confirmed by genetic studies. DiGeorge syndrome is suspected in the field of cardiac abnormalities, especially different conotruncal, hypocalcemia, cleft lip or palate and also a severe immune deficiency in infants [4-6].
Conclusion
However, the incidence of neurological symptoms along with hypocalcemia comes with a wide range of diseases as a differential diagnosis, but experts in pediatric should check cardiac disorders for the presence of DiGeorge syndrome through physical examinations and if necessary with imaging.
Conflict of Interest
The author declares that there is no conflict of interest.
Financial Support
None.
| References | ▴Top |
- Kobrynski LJ, Sullivan KE. Velocardiofacial syndrome, DiGeorge syndrome: the chromosome 22q11.2 deletion syndromes. Lancet. 2007;370(9596):1443-1452.
doi - Gonzalez W, Bautista RE. Seizures and EEG findings in an adult patient with DiGeorge syndrome: a case report and review of the literature. Seizure. 2009;18(9):648-651.
doi pubmed - Al-Jenaidi F, Makitie O, Grunebaum E, Sochett E. Parathyroid gland dysfunction in 22q11.2 deletion syndrome. Horm Res. 2007;67(3):117-122.
pubmed - Doyle DA. Hyperparathyroidism. In: Kliegman RM, Stanton B, Geme JS, Schor NF, Behrman RE. Nelson textbook of pediatrics. Elsevier Health Sciences; 2015: p. 2690-2693.
- Kliegman RM, Stanton B, Geme JS, Schor NF. Cyanotic Heart Lesions: Lesions associated with decreased pulmonary blood flow. In: Kliegman RM, Stanton B, Geme JS, Schor NF, Behrman RE. Nelson textbook of pediatrics. Elsevier Health Sciences; 2015: p. 2211-2222.
- Bringhurst FR, Demay MB, Kronenberg HHM. Hormones and disorder of mineral metabolism. In: Melmed S, Polonsky KS, Larsen PR, Kronenberg HM. Williams textbook of endocrinology. Elsevier Williams Text book Oer He2th Sciences; 2012: p. 1203-1242.
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Neurology Research is published by Elmer Press Inc.