

| Journal of Neurology Research, ISSN 1923-2845 print, 1923-2853 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Neurol Res and Elmer Press Inc |
| Journal website http://www.neurores.org |
Case Report
Volume 9, Number 4-5, October 2019, pages 81-88
Characteristic Analysis and Literature Review of Hereditary Spinocerebellar Ataxia With Lumbar Spondylolisthesis and Valvular Prolapse
Yi Baoa, b, Wanjuan Tanga, Siqin Zhoua, Ying Wanga, Jing Xiaoa, Lei Gaoa, Ran Ana, Guangjian Liua
aDepartment of Neurology, Taihe Hospital Affiliated to Hubei University of Medicine, Shiyan City, Hubei, China
bCorresponding Author: Yi Bao, Department of Neurology, Taihe Hospital Affiliated to Hubei University of Medicine, Shiyan City, Hubei, China
Manuscript submitted June 13, 2019, accepted July 30, 2019
Short title: Characteristic of SCA with Valvular Prolapse
doi: https://doi.org/10.14740/jnr544
| Abstract | ▴Top |
Spinocerebellar ataxia (SCA) is an autosomal dominant disease with high genetic heterogeneity, which cannot be cured until now. According to clinical manifestations or genetic pathology classification, SCA types 1 to 47 have been characterized so far, and the pathogenic genes of 28 SCA types have been identified. The clinical manifestations of the disease are diverse and easy to be misdiagnosed. This study aims to describe the family characteristics, specific clinical manifestations and genotyping of SCA patients. Magnetic resonance imaging (MRI) was used to check the atrophy of the brain and spinal cord. The lumbar spondylolisthesis was examined by computed tomography (CT). The possible influencing factors were analyzed by questioning each member of the patient’s family, especially the persons with the disease, to draw the genetic genealogy. Relevant literatures were searched to compare differences in genotypes between the patient and similar clinical manifestations. Craniocerebral MRI showed that cerebellar sulcus widened and deepened, vermis atrophy; enlargement of the cistern around the brainstem; cerebral cortex atrophy, furrow, fissure widen. Lumbar CT showed L3 spondylolisthesis slightly to the right. Genetic genealogy showed that the children of the patients still had the disease, and the children of the patients without the disease were all normal, which is consistent with the autosomal dominant genetic law. Compared with the literature, the patient had the same clinical manifestations as Machado’s disease: convex eyes, dysarthria, terminal muscles atrophy, ataxia gait, weakened tendon reflex, and arched foot. The same clinical manifestations of SCA40 included ataxia, wide-based gait, poor range discrimination and rotation movement disorder, but there were also many discrepancies. The patient’s lumbar spondylolisthesis and valvular prolapse were not present in any of the previous types. In conclusion, craniocerebral MRI and gene sequencing can help distinguish and diagnosis the subtypes of SCA; whether this patient is a new subtype with lumbar spondylolisthesis and heart valve prolapse needs further study; this genealogy supports that through eugenics dominant genetic diseases being passed on to the offspring can be avoided.
Keywords: SCA; Lumbar spondylolisthesis; Valvular prolapse; Machado; Genealogy
| Introduction | ▴Top |
Spinocerebellar ataxia (SCA) lesions often involve the spinal cord, cerebellum and brainstem, which are autosomal dominant genetic diseases with high genetic heterogeneity, with usually 30 - 40 years old insidious onset and slow progress. There is childhood or over 70 years old onsets; and most of them become unable to walk 10 - 20 years after the onset. Ataxia of lower limbs is almost the first symptom of SCA, which is manifested as unstable gait, fall easily, unclear speech, clumsy hands, intention tremor, nystagmus, dementia and distal muscular atrophy. Physical examination shows dystonia, hyperreflexia, pathological positive, spasmodic gait, tuning fork vibration and body sensation loss. The clinical manifestations are diverse, and there are no effective treatments to date [1, 2].
With the development of medical genetics and the extensive clinical application of next-generation sequencing, SCA is divided into subtypes such as SCA type 1 to SCA type 47 according to the differences of related chromosomes, gene loci, coding products and clinical manifestations [3-11]. The distribution of SCA subtypes is different in different countries and races. Among them, SCA3 (Machado-Joseph disease, MJD) is the commonest subtype of Chinese gene detection, accounting for more than 50% of SCA patients. The gene is located on the chromosome 14q32.1 and encodes the ataxin-3 (ATXN3) protein [12]. The clinical manifestations of SCA3 are diverse, mainly characterized by progressive cerebellar ataxia; and some patients are mainly characterized by pyramidal tract signs (muscle atrophy, abnormal muscle tone) and extrapyramidal signs (Parkinson’s disease, chorea), and even spastic paraplegia and other clinical symptoms [4]. SCA40 gene is located at 14p32.11 and can encode the protein CCDC88C, which is one of the SCA type newly discovered. It is mainly manifested as ataxia, wide-based gait, poor distance distinguish, intention tremor, rotation movement disorder, and tendon reflex hyperactivity. The age of the onset is generally greater than 42 years [9]. In general, patients often suffer from unstable walking as the first symptom, mistakenly believing that it is a lumbar disease, and seek medical treatment in the Spinal Surgery.
Lumbar spondylosis is clinically manifested as lumbago and limited lumbar activity, which is caused by acute and chronic injury of the spine and soft tissue around the spine, lumbar disc degeneration, and lumbar vertebrae hyperplasia. Due to that the location and size of nucleus pulposus protrusion, spinal canal diameter, pathological characteristics, body state and individual sensitivity are different, the clinical manifestations differ. In general, patients present with walking instability, weakened tendon reflex, terminal muscular atrophy, and other problems, similar to the clinical manifestations of spinal cerebellar ataxia [13].
In this study, the patient’s walking instability was the most painful manifestation, accompanied by lumbago. Lumbar spine magnetic resonance imaging (MRI) and computed tomography (CT) examination in the Spine Surgery Department revealed lumbar disc herniation and spondylolisthesis, which was diagnosed as lumbar spondylolisthesis. After treatment for the lumbar vertebrae, the patient’s symptoms did not improve, and he was transferred to the Department of Neurology. Through detailed neurological examination and questioning of the family history, the final diagnosis of spinal cerebellar ataxia was proposed; but the patient refused to undergo gene examination, so we could not know the accurate genotype. After literature review, it was compared with the clinical manifestations of SCA type 1 to SCA type 47 in detail, which revealed that this patient were highly similar to those of SCA3 and SCA40; but there were also a variety of manifestations inconsistent with them, and the lumbar spondylolisthesis and valve prolapse of this patient were not the manifestations of the previous types.
| Case Report | ▴Top |
A 61-year-old male patient was admitted to the Spine Surgery Department due to unsteady walking for 3 years without previous special medical history. Lumbar MRI examination showed that L4-S1 vertebral body relative edge saw strip T2WI high signal, lumbar intervertebral disc T2WI signal decreased, L3-4 intervertebral disc protruded to the left rear, L4-5 intervertebral disc protruded to the right rear, L5-S1 intervertebral disc protruded to the posterior aspect, dural sac was compressed and deformed, bilateral crypt was slightly narrow, and nerve root was slightly compressed; Lumbar CT revealed that L3 vertebral body slightly shifted to the right, which was diagnosed as lumbar spondylolisthesis. After lumbar external fixation, physiotherapy, massage, acupuncture and moxibustion, improving microcirculation therapy and other treatments, the patient did not get better, and he was transferred to the Department of Neurology after consultation. His medical history showed that in the past 3 years, the patient had unstable gait, fell easily, the tone of speech changed, occasionally drinking water could easily lead to coughing, limbs muscles gradually atrophied; and there were many people in the family that had similar presenting symptoms in the same age group.
Study methods
Draw genetic genealogy map
There were many members of the proband family, which could be traced back to the previous two generations and the next generation. Through the description by the proband and telephone inquiry to the family members, the relationship between the family members and the proband, clinical manifestations, age of onset, progress and final results were recorded in detail, and the genetic genealogy map was drawn.
Neurological examination
The patient was conscious, proptosis, both pupils equally large and round, with a diameter of 3 mm, sensitive to light reflection, freely moving in all directions of the eyeballs on both sides, without nystagmus; facial muscle atrophy, bilateral nasolabial groove symmetry, speech tone abnormalities, drinking water could easily lead to coughing, hearing loss; the muscles of the limbs were atrophic, the muscle strength and muscle tension were normal, decreased knee and Achilles tendon reflexes, deep and superficial sensation without abnormalities, finger-nose test was not accurate, poor ability to discriminate distance, rotation test positive, Romberg sign (+), wide-based gait, heel-knee-shin test positive, arched foot; neck soft and no resistance, bilateral Kernig’s sign and Brudzinski’s sign all negative.
Diagnosis and treatment
According to the patient’s symptoms, signs and family history, the diagnosis of hereditary spinocerebellar ataxia was considered. Relevant examinations were performed: craniocerebral MRI, cervical MRI, brainstem auditory evoked potential (BAEP), fundus examination, limb electromyography (EMG), electrocardiogram (ECG), cardiac color Doppler ultrasound, blood routine, thyroid function, etc. He was suggested to perform the family gene and neurological pathology examination, but the patient refused. When the patient learned that there was no effective treatment for the disease, he required to be discharged.
Relevant literature retrieval and analysis
We searched related original papers from PubMed, Medline, ISI Web of Science, Elsevier Science Direct, China Knowledge Network (CNXI), Chongqing Weipu (VIP) and Wanfang, etc. The retrieval time was from the database construction to May 1, 2019, and the keywords used are: spinocerebellar ataxia, SCA, Machado-Joseph disease, MJD, CCDC88C, and genetic genealogy map. The search method was the subject words combined with free words. In addition, references from qualified publications were manually screened for potential references. Inclusion and exclusion criteria: duplicate references were deleted. During title and abstract screening, literature that did not meet our inclusion criteria was excluded. Genetic loci, coding proteins, CAG repeats, age of onset, clinical manifestations and complications of each SCA type and subtype were collected according to literature. The subtypes with similar clinical symptoms in this patient were tabulated to compare their genetic and clinical heterogeneity.
Study results
Routine inspection
BAEP showed extensive bilateral lesions consistent with the patient’s hearing loss. NCV showed that the conduction velocity and amplitude of femoral nerve, tibial nerve, common peroneal nerve, lateral femoral cutaneous nerve and superficial peroneal nerve were normal. EMG showed that no significant fibrillation and positive sharp wave in the rectus femoris, tibialis anterior and gastrocnemius; the unit amplitude and time limit of light contraction motion were normal; it indicated that the patient’s lower limb nerves and muscles had no obvious damage. Blood routine showed: hemoglobin 98 g/L, hematocrit 0.319, average red blood cell volume 67.9 fL, average hemoglobin concentration 307 g/L, suggesting mild small-cell hypochromic anemia resulted from iron deficiency. Vitamin examination showed that vitamin D 22.46 nmol/L, vitamin E 9.38 µg/mL, vitamin B1, vitamin B6 and vitamin B12 were not significantly abnormal, which could exclude neuropathy caused by vitamin metabolic disorders, such as spinal cord subacute combined degeneration. The patient had normal thyroid function and the associated metabolic disease might not be considered.
Craniocerebral and cervical MRI
The results of craniocerebral MRI showed cerebellar sulcus widened and deepened, vermis atrophy, enlargement of the brain pool around the brain stem indicated that the brain stem became smaller, cerebral cortex atrophy, furrow and fissure widen. Cervical MRI showed that the cervical spinal cord was significantly thinner, the surrounding space was enlarged, there was no abnormal signal in the spinal cord, there was no obvious degenerative change in the cervical spine, and the intervertebral disc was slightly herniated. Therefore, relevant neurological symptoms caused by spinal cord compression could be excluded (Fig. 1).
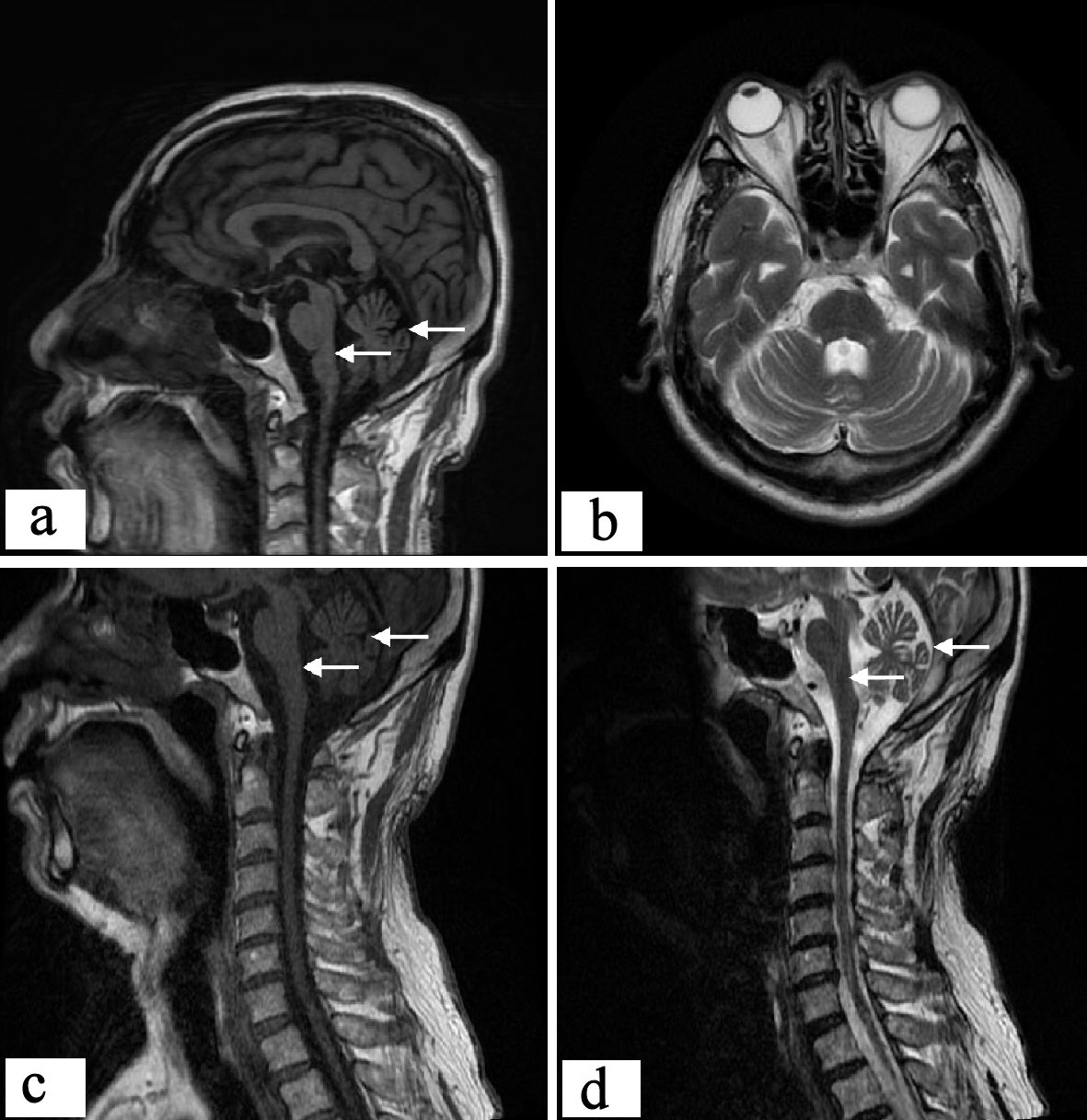 Click for large image | Figure 1. Proband’s craniocerebral and cervical MRI. Sagittal scan of craniocerebral showed: cerebellar cortical sulcus widened, cerebellar atrophy as a whole; brain stem atrophy obviously, and dorsal curvature of the pons disappeared basically; cerebellar upper pool, cerebellar medullary pool, and fourth ventricle were enlarged. Transverse craniocerebral scan showed: the cerebellar sulcus widened and deepened, and vermis atrophy; brain pool around the brain stem expands. The sagittal position of the cervical spinal cord showed: the cervical spinal cord was significantly thinner. MRI: magnetic resonance imaging. |
Cardiac examination result
The patient denied any history of chest pain. On auscultation, non-ejective clicks could be heard in the middle and late stage of apex contraction, which was considered to be caused by the chordae that was suddenly tightened or the prolapse of the leaflets suddenly stopped. It was consistent with the performance of valve prolapse. ECG examination revealed that P wave: (-), QRS group: V1 lead was rsR’ type, other lead ends were blunt, QRS total time limit > 0.11 s, ST segment: V1 - V3 lead down (secondary change), T wave: V1 - V3 lead inverted (secondary change), indicating ST segment changed and conduction blocked. Cardiac color Doppler ultrasonography showed aortic sinus and ascending aorta widened, left heart enlargement, left ventricular wall thickening, mitral valve posterior valve prolapse (the apex of the posterior flap P2-P3 region was “fishhook” took off to the left atrium, opened well) and slightly closed incomplete, left ventricular diastolic dysfunction. According to this, it could be diagnosed as valve prolapse.
Lumbar CT and MRI examination
The patient walked unsteadily, used a cane with one hand for a long time, and the center of gravity was tilted to the right. The CT examination of the lumbar coronal position showed that the lumbar vertebra 3 was slightly shifted to the right. According to Meyerding method, it accorded with degree I (slippage < 25%). Lumbar sagittal MRI showed: L3 mild posterior slippage, and disc herniation, dural sac was compressed and deformed, nerve root was slightly compressed, chondritis occurred at the opposite edge of the L4-S1 vertebral body. However, there was no direct relationship between lumbar spine lesion and walking instability (Fig. 2).
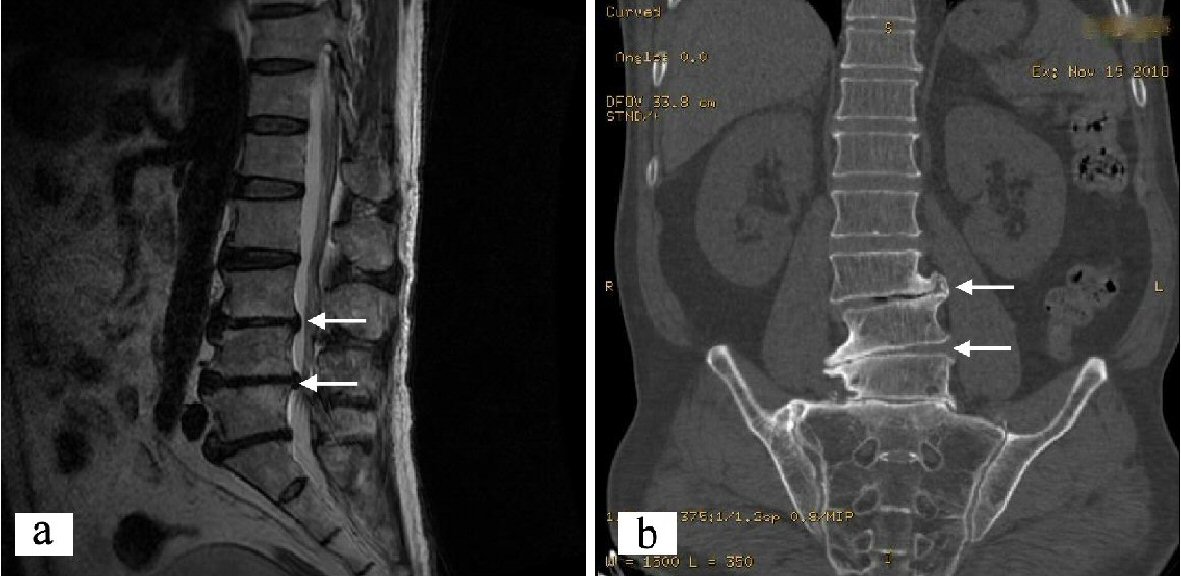 Click for large image | Figure 2. Proband’s lumbar spondylolisthesis images. (a) Lumbar sagittal MRI: L3 slipped slightly backward. (b) Lumbar CT surface reconstruction image: L3 spondylolisthesis to the right. CT: computed tomography. |
Family disease spectrum
There were 57 people with blood relationship in the family, including five patients, two suspected patients, four females and three males. All of them had symptoms around the age of 60. The symptoms were characterized by tone change, unstable walking, and widening of the walk base, muscle atrophy, and progressive aggravation. Currently, four people had died. According to the proband’s narration: the grandmother appeared gait instability around the age of 60, walking sway, and gradually paralyzed in bed, and had died; the elder aunt appeared needing to rely on walking sticks around the age of 60, and had lower limb muscle atrophy, and had died. The mother’s symptoms appeared at 63 years old, including that drinking water could easily lead to coughing, hearing loss, walking instability, easy to fall; she could not take care of herself, and limb muscle atrophy was obvious, and had died. The younger uncle’s symptoms appeared at 55 years old with progressive instability of walking, wide-based gait, crutch holding, and muscular atrophy of both lower limbs. Symptoms of the older sister (the daughter of the aunt) appeared at 60 years old with walking unsteadily and gradually becoming worse. At present, she needed to be helped go up and down stairs, occasionally falling, and the fine movements were poor. Symptoms of the younger brother (the son of the younger uncle) appeared around 50 years old, with unstable walking, and his step base was widened. However, he died unexpectedly, specific reasons unknown. The children of the sick members had not yet reached the age of 50, and there were currently no cases. The specific family relationship was shown in Figure 3.
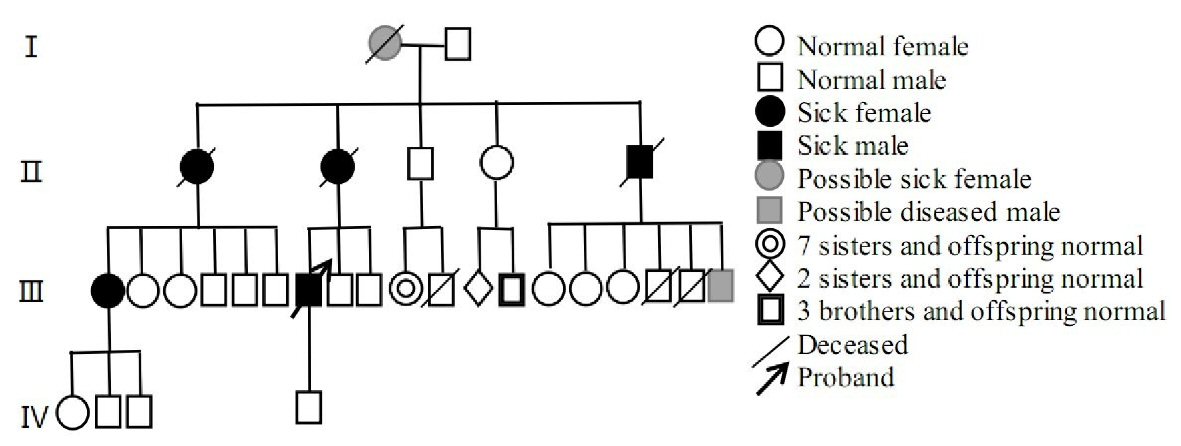 Click for large image | Figure 3. Family genealogy. |
Comparison of proband’s symptoms with SCA subtypes
According to the clinical manifestations of the patient, genotypes with similar clinical manifestations were extracted from previous literatures; and gene loci, coding proteins, CAG repetition times, age of onset, and clinical characteristics were listed, as shown in Table 1 for details [4, 10, 12, 14-18]. According to Table 1, the patient had proptosis, facial muscle atrophy, hearing loss, tone anomaly, drinking water could easily lead to coughing, limb muscle atrophy, muscle strength and muscle tension normal, wide-based gait, decreased knee and Achilles tendon reflexes, deep and superficial sensation without abnormalities, finger-nose test was not accurate, poor ability to discriminate distance, rotation test positive, Romberg sign (+), heel-knee-shin test positive, arched foot, and lumbar spondylolisthesis; which was Similar to Machado’s disease [4, 17, 18] and SCA40 [9, 19]. Compared with Machado’s disease, there was no soft palate weakness, nystagmus, eyelid opening difficulty, upper vision difficulty, limb bundle tremor, scoliosis or hypoesthesia of vibration. Compared with type SCA40, there was no intention tremor or tendon hyperreflexia.
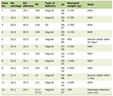 Click to view | Table 1. Genetic and Clinical Features of Similar SCA Subtypes |
| Discussion | ▴Top |
SCA is a large class of neurodegenerative degenerative diseases characterized by chronic progressive ataxia, a genetic defect caused by abnormal doubling mutations in CAG nucleic acid repeats, which repeatedly encodes the polyglutamine channel on the P/Q type calcium channel alpha 1A subunit. In many cases the amplified fragment size is associated with disease severity. The diverse clinical manifestations are mainly associated with the damage and dysfunction of cerebellum, pyramidal system, extrapyramidal system, motor neurons and other systems. Symptoms of pyramidal system include muscular atrophy, dystonia, and even spastic paraplegia. Extrapyramidal symptoms may also occur such as Parkinson’s disease and chorea. A few patients only have extrapyramidal symptoms, with cerebellar disorders symptoms not obvious, especially in the early stage. It is difficult to distinguish from those diseases with similar clinical symptoms, such as lumbar spondylosis, Parkinson’s disease, chorea, and bradykinesia, and it is easy to be misdiagnosed [2, 5, 11, 14].
The current clinical diagnosis of spinal cerebellar ataxia is based on: 1) Ataxia, dysarthria, pyramidal tract signs and other typical common symptoms, which can be accompanied by ophthalmic palsy, extrapyramidal symptoms and retinal pigmentation; 2) MRI examination found cerebellum, brain stem atrophy, and excluded other involvement of cerebellum and brain stem degeneration; 3) Other related auxiliary examinations such as X-ray showed spine and bone malformations, brain stem evoked potential could be abnormal, EMG showed peripheral nerve damage, ECG showed common T wave inversion, arrhythmia and conduction block, echocardiography showed ventricular hypertrophy, and visual evoked potential amplitude decreased. Clinical diagnosis is only made according to the characteristic symptoms and signs of each subtype, but it is difficult to make an accurate classification (except SCA7). At present, the second-generation sequencing method can be used to accurately determine the typing at the molecular level and detect the number of CAG amplifications [2, 11, 12, 15].
Autosomal dominant genetic disease is characterized by a single allele mutation that can cause the disease, equal opportunities for men and women; and 50% of offspring have the possibility of developing the disease. Common subtypes include: 1) Complete dominance; 2) Incomplete dominance; 3) Irregular dominant; 4) Common explicit; 5) Delay dominance; 6) Sex-influenced dominance. In a patient’s family, the disease can occur for several generations, but sometimes due to changes in the internal and external environment, the role of the causative gene may not be expressed (explicit incomplete). In the family of the proband, the elder aunt, mother and younger uncle all had the disease, and their children also had the disease. Elder uncle and younger aunt were normal, and after marriage with normal people, their offspring had no disease, which was consistent with autosomal dominant heredity rule [1, 9, 10, 16].
The patient’s clinical features included: proptosis, facial muscle atrophy, hearing loss, tone anomaly, drinking water could easily lead to coughing, limb muscle atrophy, muscle strength and muscle tension normal, wide-based gait, decreased knee and Achilles tendon reflexes, deep and superficial sensation without abnormalities, finger-nose test was not accurate, poor ability to discriminate distance, rotation test positive, Romberg sign (+), heel-knee-shin test positive, arched foot, and lumbar spondylolisthesis. According to reports in the literature, proptosis is common in SCA3, dysarthria is found in SCA3, SCA20, SCA23, SCA30, muscle atrophy is seen in SCA3, SCA18, weakened tendon reflexes is seen in SCA2, SCA3, SCA4, SCA19, SCA22, SCA25, and heart damage is seen in SCA7 [3, 4, 10, 12, 20-23]. SCA3 is also known as Machado-Joseph disease, which is mainly characterized by cerebellar ataxia, different degrees of pyramidal tract signs, extrapyramidal signs or peripheral muscle atrophy. In which the type III also called Machado disease accounted for 47%, with the onset after 50 to 60 years old, and with cerebellar dysfunction and motor neuropathy (symmetric extremity muscle atrophy) is given priority to, also may be accompanied by extraocular muscle paralysis and pyramidal tract, slow progress; the prognosis is good. Compared with Machado’s disease, the patient had proptosis dysarthria, extremity muscle atrophy, ataxia gait, weakened tendon reflex and arched foot, but no nystagmus, eyelid opening difficulty, upper vision difficulty, soft palate weakness, limb bundle tremor, decreased vibration sensation, peripheral neuropathy or scoliosis. More importantly, there is no obvious genetic anticipation predisposition in the family of the proband [17, 18, 23]. SCA40 is one of the newly discovered SCA subtype. The gene is located at 14p32.11, which can encode CCDC88C protein. The age of onset is generally greater than 42 years old, mainly manifested as ataxia, accompanied by broad basal gait, poor distance discrimination, intention tremor, rotation movement disorder, and tendon hyperreflexia. Compared with SCA40, although the patient has ataxia, accompanied by broad basal gait, poor distance discrimination, rotation movement disorder, but there is no intention tremor, or tendon hyperreflexia [9, 10, 12, 19].
In addition to the above-mentioned performance, the patient also had clinical features not shown in SCA1-40, with lumbar spondylolisthesis apparent, but no lumbar scoliosis consistent with Machado’s disease. According to the patient with abnormal gait and walking on crutch for a long time, it is considered that lumbar spondylolisthesis was a displacement caused by uneven spinal column force resulting in abnormal bone connection of adjacent vertebral bodies. CT showed partial slip of the upper vertebral body and the lower vertebral body, but the lumbar involvement of the disease should also be considered. Although the patient denied the history of chest pain and palpitation, physical examination showed abnormal valve murmur. ECG showed: total QRS time limit > 0.11 s, ST segment: V1 - V3 leads down (secondary change), T wave: V1 - V3 lead inverted (secondary change); cardiac ultrasonography showed: mitral valve posterior valve prolapse (the apex of the posterior flap P2-P3 region was “Fishhook” took off to the left atrium, opened well), which was consistent with the cardiac involvement of SCA7. Mitral valve prolapse syndrome is a congenital connective tissue disease, commonly seen in women aged 14 to 30 years old. The symptoms are mostly non-specific, which can be manifested as chest pain, palpitations and arrhythmia in the precardiac area. About one-fourth to one-fifth of the patients have no symptoms. This patient had valve prolapse, whether it is the manifestation of cardiac damage of the disease, has not been reported in the literature.
The reasons for misdiagnosis of this case are as follows: 1) SCA is a rare disease, and the public lacks relevant knowledge, so it is often treated in other departments; 2) The patient was diagnosed as lumbar spondylosis due to late onset, long-term abnormal posture, walking swing, severe lumbar spine damage, and combined with degenerative changes of lumbar spine. 3) The clinical manifestations of this disease are diverse. The general doctor did not conduct a detailed neurological examination, and did not pay attention to the family history, which is likely to lead to the misdiagnosis.
Current treatment progress is that once the disease is onset, there is no specific treatment; and symptomatic treatment is often used to relieve symptoms. In terms of drug therapies, amantadine can improve ataxia; chloranilide can reduce spasm; levodopa can relieve stiffness and other extrapyramidal symptoms; ataxia with myoclonus preferred clonazepam, ATP, coenzyme A; inosine and vitamin B family are used to nourish nerves; butylphthalide and idebenone have protective mitochondria and antioxidant functions. If the drug treatment is difficult to relieve the symptoms, it is feasible that the optic thalamus be destroyed. In addition, rehabilitation training, physical therapy and assisted walking instruments can improve limb function and avoid posture abnormalities leading to lumbar spondylolisthesis [2, 10-12, 19].
There are some shortcomings of this article. 1) There is no genetic testing of the whole family, so it is difficult to make a diagnosis at the gene level, and only clinical diagnosis can be made; 2) The patient did not complete neuropathological examination, which is one of the defects of this case; 3) This case is still difficult to be diagnosed as a certain subtype at present. According to clinical manifestations, signs, lumbar spondylolisthesis and valve prolapse, whether a new subtype can be considered. But the sample size is small, more sample size needs to be accumulated to support it. 4) Young members of the patient family have not yet reached the age of onset, which requires long-term follow-up.
Conclusions
SCA is an autosomal dominant genetic disease, after onset, the patient’s condition is progressively aggravated. Currently, there is no effective treatment method, which seriously reduces the quality of life, and it requires long-term care of the family members. In order to avoid this disease, premarital genetic screening, prenatal diagnosis and genetic counseling are recommended to reduce the birth rate of child patient. If there are similar symptoms in the family, early genetic diagnosis and drug intervention should be carried out to improve the quality of life. Establish family case database and biological sample database is important to promote the current research progress. In recent years, a variety of biological functions of polyQ SCA genes/proteins, including transcription, RNA splicing and metabolic regulation, and deubiquitinating enzyme activities have made considerable progress, helping to determine the pathogenesis of each disease, which may become a potential target for the treatment of motor dysfunction. Use of current CRISPR/Cas9 technology may perform gene editing, remove pathogenic genes, and improve the eugenics rate.
Acknowledgments
We are grateful to the medical staff who have treated those patients carefully.
Financial Disclosure
This article is a clinical observation study, no funding.
Conflict of Interest
We declare no conflict of interest.
Informed Consent
Not applicable.
Author Contributions
Data collection: YB, WJT, SQZ; data analysis: YB, JX, LG; manuscript preparation: YB, YW, WJT, SQZ; reviewing of manuscript: YB, RA, GJL.
| References | ▴Top |
- Ozaki K, Doi H, Mitsui J, Sato N, Iikuni Y, Majima T, Yamane K, et al. A novel mutation in ELOVL4 leading to Spinocerebellar Ataxia (SCA) with the hot cross bun sign but lacking erythrokeratodermia: a broadened spectrum of SCA34. JAMA Neurol. 2015;72(7):797-805.
doi pubmed - Tsai YA, Liu RS, Lirng JF, Yang BH, Chang CH, Wang YC, Wu YS, et al. Treatment of Spinocerebellar Ataxia With Mesenchymal Stem Cells: A Phase I/IIa Clinical Study. Cell Transplant. 2017;26(3):503-512.
doi pubmed - Chong SS, McCall AE, Cota J, Subramony SH, Orr HT, Hughes MR, Zoghbi HY. Gametic and somatic tissue-specific heterogeneity of the expanded SCA1 CAG repeat in spinocerebellar ataxia type 1. Nat Genet. 1995;10(3):344-350.
doi pubmed - Jacobi H, du Montcel ST, Bauer P, Giunti P, Cook A, Labrum R, Parkinson MH, et al. Long-term disease progression in spinocerebellar ataxia types 1, 2, 3, and 6: a longitudinal cohort study. Lancet Neurol. 2015;14(11):1101-1108.
doi - Trang H, Stanley SY, Thorner P, Faghfoury H, Schulze A, Hawkins C, Pearson CE, et al. Massive CAG repeat expansion and somatic instability in maternally transmitted infantile spinocerebellar ataxia type 7. JAMA Neurol. 2015;72(2):219-223.
doi pubmed - Nethisinghe S, Lim WN, Ging H, Zeitlberger A, Abeti R, Pemble S, Sweeney MG, et al. Complexity of the genetics and clinical presentation of spinocerebellar ataxia 17. Front Cell Neurosci. 2018;12:429.
doi pubmed - Stevanin G, Broussolle E, Streichenberger N, Kopp N, Brice A, Durr A. Spinocerebellar ataxia with sensory neuropathy (SCA25). Cerebellum. 2005;4(1):58-61.
doi pubmed - Corral-Juan M, Serrano-Munuera C, Rabano A, Cota-Gonzalez D, Segarra-Roca A, Ispierto L, Cano-Orgaz AT, et al. Clinical, genetic and neuropathological characterization of spinocerebellar ataxia type 37. Brain. 2018;141(7):1981-1997.
doi pubmed - Tsoi H, Yu AC, Chen ZS, Ng NK, Chan AY, Yuen LY, Abrigo JM, et al. A novel missense mutation in CCDC88C activates the JNK pathway and causes a dominant form of spinocerebellar ataxia. J Med Genet. 2014;51(9):590-595.
doi pubmed - Sullivan R, Yau WY, O'Connor E, Houlden H. Spinocerebellar ataxia: an update. J Neurol. 2019;266(2):533-544.
doi pubmed - Diallo A, Jacobi H, Cook A, Labrum R, Durr A, Brice A, Charles P, et al. Survival in patients with spinocerebellar ataxia types 1, 2, 3, and 6 (EUROSCA): a longitudinal cohort study. Lancet Neurol. 2018;17(4):327-334.
doi - Chen Z, Wang P, Wang C, Peng Y, Hou X, Zhou X, Li T, et al. Updated frequency analysis of spinocerebellar ataxia in China. Brain. 2018;141(4):e22.
doi pubmed - Fortin M, Lazary A, Varga PP, Battie MC. Association between paraspinal muscle morphology, clinical symptoms and functional status in patients with lumbar spinal stenosis. Eur Spine J. 2017;26(10):2543-2551.
doi pubmed - Sun YM, Lu C, Wu ZY. Spinocerebellar ataxia: relationship between phenotype and genotype - a review. Clin Genet. 2016;90(4):305-314.
doi pubmed - Fogel BL, Lee H, Deignan JL, Strom SP, Kantarci S, Wang X, Quintero-Rivera F, et al. Exome sequencing in the clinical diagnosis of sporadic or familial cerebellar ataxia. JAMA Neurol. 2014;71(10):1237-1246.
doi pubmed - Schols L, Bauer P, Schmidt T, Schulte T, Riess O. Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol. 2004;3(5):291-304.
doi - Jacobi H, Bauer P, Giunti P, Labrum R, Sweeney MG, Charles P, Durr A, et al. The natural history of spinocerebellar ataxia type 1, 2, 3, and 6: a 2-year follow-up study. Neurology. 2011;77(11):1035-1041.
doi pubmed - Jacobi H, Reetz K, du Montcel ST, Bauer P, Mariotti C, Nanetti L, Rakowicz M, et al. Biological and clinical characteristics of individuals at risk for spinocerebellar ataxia types 1, 2, 3, and 6 in the longitudinal RISCA study: analysis of baseline data. Lancet Neurol. 2013;12(7):650-658.
doi - Paulson HL, Shakkottai VG, Clark HB, Orr HT. Polyglutamine spinocerebellar ataxias - from genes to potential treatments. Nat Rev Neurosci. 2017;18(10):613-626.
doi pubmed - Knight MA, Gardner RJ, Bahlo M, Matsuura T, Dixon JA, Forrest SM, Storey E. Dominantly inherited ataxia and dysphonia with dentate calcification: spinocerebellar ataxia type 20. Brain. 2004;127(Pt 5):1172-1181.
doi pubmed - Serrano-Munuera C, Corral-Juan M, Stevanin G, San Nicolas H, Roig C, Corral J, Campos B, et al. New subtype of spinocerebellar ataxia with altered vertical eye movements mapping to chromosome 1p32. JAMA Neurol. 2013;70(6):764-771.
doi pubmed - Schelhaas HJ, van de Warrenburg BP. Clinical, psychological, and genetic characteristics of spinocerebellar ataxia type 19 (SCA19). Cerebellum. 2005;4(1):51-54.
doi pubmed - Linnemann C, Tezenas du Montcel S, Rakowicz M, Schmitz-Hubsch T, Szymanski S, Berciano J, van de Warrenburg BP, et al. Peripheral neuropathy in spinocerebellar ataxia type 1, 2, 3, and 6. Cerebellum. 2016;15(2):165-173.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Neurology Research is published by Elmer Press Inc.