

| Journal of Neurology Research, ISSN 1923-2845 print, 1923-2853 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Neurol Res and Elmer Press Inc |
| Journal website http://www.neurores.org |
Case Report
Volume 10, Number 1, February 2020, pages 13-16
Atypical Teratoid/Rhabdoid Tumor of the Sellar Region: A Case Report and Review of the Literature
Raghad Adnan Bokharia, d, Mohammed Bafaqeehb, Saad Al-Obaysib, Areej Al-Amanc, Wafa Alshakweera
aDepartment of Pathology and Clinical Laboratory Medicine, King Fahad Medical City, Riyadh, Saudi Arabia
bDepartment of Neurosurgery, National Neuroscience Institute, King Fahad Medical City, Riyadh, Saudi Arabia
cCollege of Medicine, King Saud University, Riyadh 12372, Saudi Arabia
dCorresponding Author: Raghad Adnan Bokhari, Department of Pathology and Clinical Laboratory Medicine, King Fahad Medical City, Riyadh, Saudi Arabia.
Manuscript submitted October 29, 2019, accepted November 18, 2019
Short title: A Case of Atypical Teratoid/Rhabdoid Tumor
doi: https://doi.org/10.14740/jnr556
| Abstract | ▴Top |
Atypical teratoid/rhabdoid tumors (AT/RTs) are rare and aggressive pediatric malignant rhabdoid tumors (MRTs) that occur within the brain. The majority of these tumors occur in the cerebellum. Only 45 cases of adults with AT/RTs have been reported in the literature to date. We present a case of sellar and suprasellar AT/RT in a 40-year-old female patient with this rare entity. To our knowledge, this is the 14th case of an adult-onset AT/RT in the sellar and suprasellar region.
Keywords: Atypical teratoid/rhabdoid tumor; Sellar/suprasellar lesion; Surgery
| Introduction | ▴Top |
Atypical teratoid/rhabdoid tumors (AT/RTs) are rare and aggressive pediatric malignant rhabdoid tumors (MRTs) that occur within the brain [1]. It is initially reported in 1987 and subsequently, defined as a distinct central nervous system (CNS) neoplasm in 1996. It is added to the World Health Organization (WHO) Brain Tumor Classification in 2007 (grade IV) [2, 3]. Histologically, AT/RTs are known that they are composed of diffuse proliferation of atypical large cells showing eccentrically located nuclei and abundant eosinophilic cytoplasm (rhabdoid features) with prominent nucleoli [4]. AT/RTs are characterized by biallelic loss of SMARCB1 (SWI/SNF- related, matrix-associated, actin-dependent regulator of chromatin, subfamily b, member 1). Here, we report a case of AT/RT that originated in sellar and suprasellar region in a 40-year-old female patient with brief review of this aggressive tumor in adult population.
| Case Report | ▴Top |
A medically free 40-year-old female, presented to the emergency department with a history of severe headache and right eye decrease vision. It was associated with nausea, vomiting and photosensitivity for the last few days. Clinical examination revealed decreased vision of the right eye and third nerve palsy of the same side. She denied any history of prolactinoma, constitutional symptoms or family history of pituitary tumors. Magnetic resonance imaging (MRI) and computed tomography (CT) scan showed sellar and suprasellar lesion that was invading the cavernous sinus bilaterally, but more into the right side and protruding through the sella turcica.
Preoperative MRI identified a large sellar enhancing lesion with cystic degeneration measuring 2.9 × 1.7 × 2.3 mm (Fig. 1).
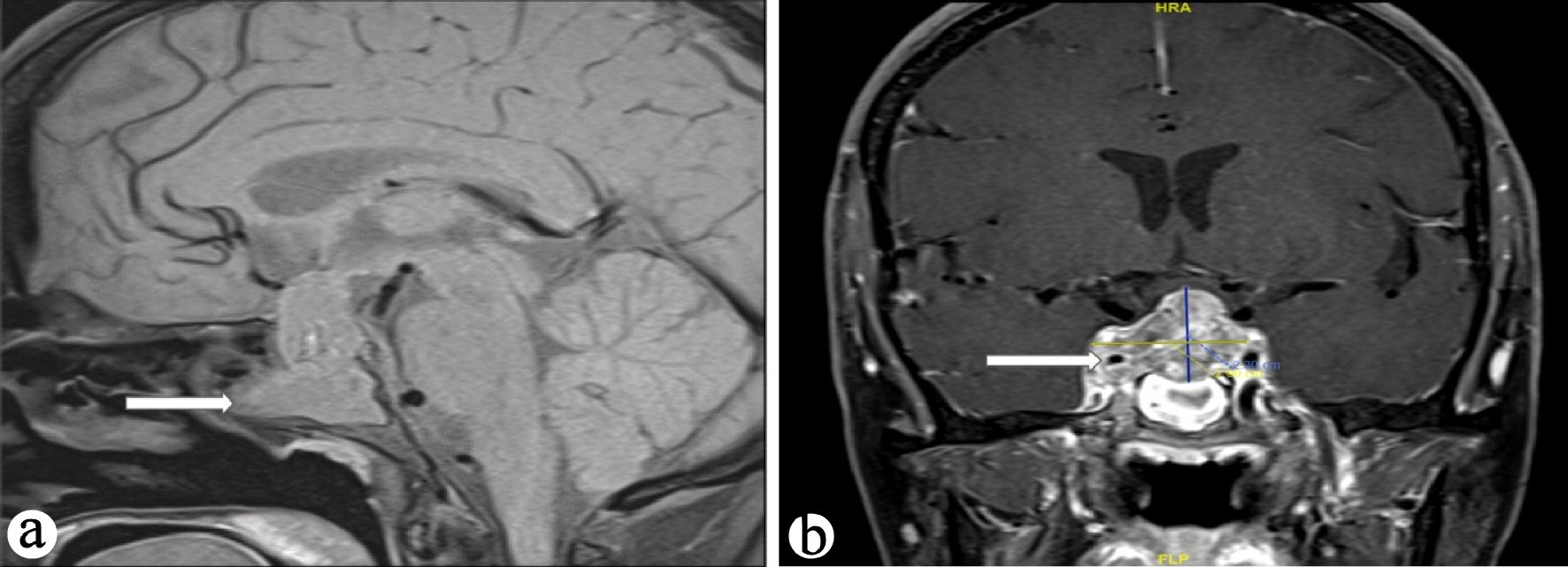 Click for large image | Figure 1. (a) T1-weighted MRI sagittal images showing large intrasellar mass (arrow), (b) T1-weighted MRI coronal images showing large intrasellar mass encasing right internal carotid artery (arrow). MRI: magnetic resonance imaging. |
Hormone profiles including cortisol, plasma adrenocorticotropic hormone (ACTH), thyroid hormones such as thyroid-stimulating hormone (TSH) and T4, follicle stimulating hormone (FSH), luteinizing hormone (LH) and growth hormone, prolactin were normal.
Classical transnasal endoscopic approach was performed with subtotal resection of the sellar and suprasellar lesion. The tumor was rubbery and fibrotic which was unusual to pituitary adenoma.
The histopathological study revealed a high-grade densely cellular neoplasm (Fig. 2). Tumor cells were large-sized, polygonal in shape and arranged in sheets. The cytoplasm was eosinophilic with focal eccentric eosinophilic globular inclusions. The nuclei were oval-shaped and pleomorphic with prominent nucleoli (Fig. 3). First differential diagnosis was pituitary adenoma, as it is the most common tumor in the sellar region. Other differential diagnoses include glioma and AT/RTs. Negative staining for pituitary panel, chromogranin and synaptophysin ruled out pituitary adenoma and other neuroendocrine tumors as well. Glial fibrillary acidic protein (GFAP) was negative excluding glial tumor. Immunohistochemical stain for INI-1 (BAF-47) was negative in the tumor nuclei (Fig. 4). The morphologic characteristics and immunoprofile were diagnostic of AT/RT.
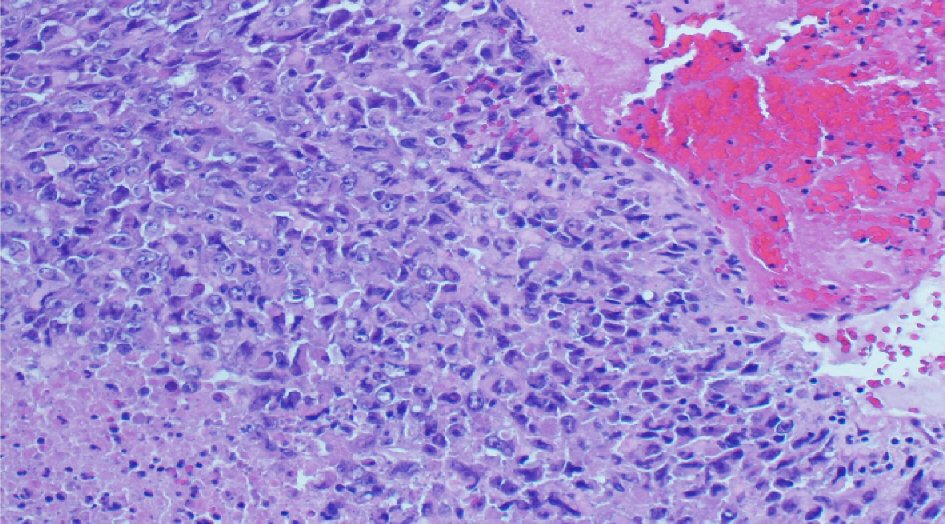 Click for large image | Figure 2. The tumor is composed of sheets of undifferentiated cells with large area of necrosis and hemorrhage (H&E stain, × 200 magnifications). H&E: hematoxylin and eosin stain. |
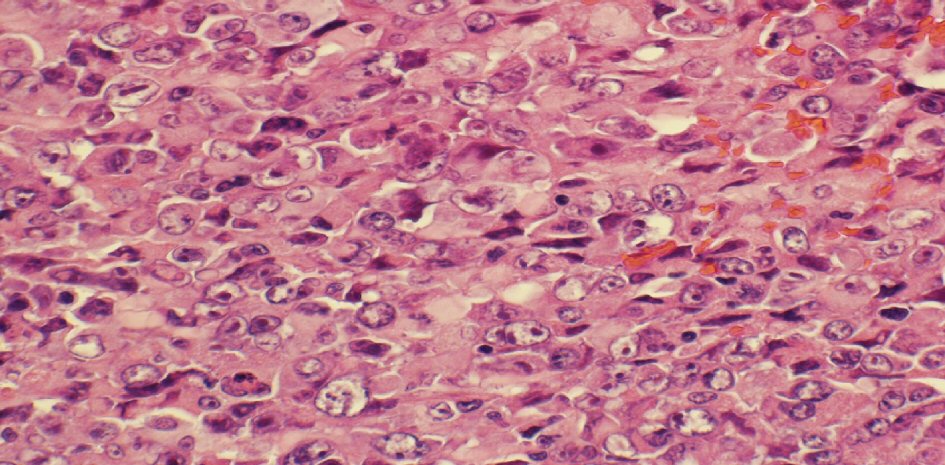 Click for large image | Figure 3. The tumor is composed of medium sized to large cells with prominent nucleoli and some with peripherally placed nuclei (H&E stain, × 400 magnifications). H&E: hematoxylin and eosin stain. |
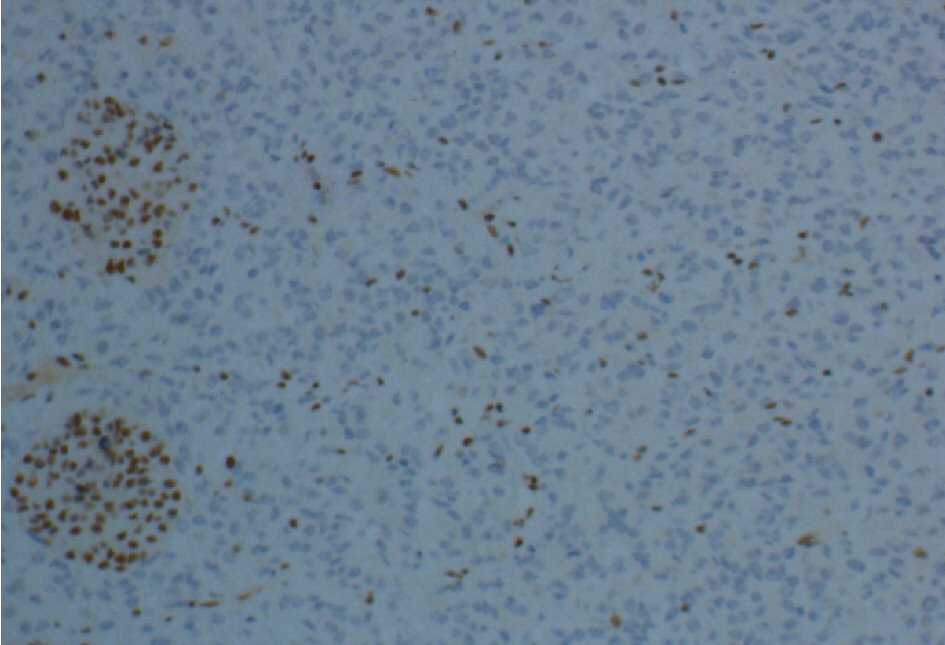 Click for large image | Figure 4. INI-1 (BAF-47) immunostaining shows loss of nuclear staining in the tumor nuclei and positive in the endothelial cells and residual entrapped pituitary tissue (× 200 magnification). |
Postoperatively, the patient developed diabetes insipidus (DI), which was treated by desmopressin. After that, she was referred to oncology team and started on chemoradiation therapy. One month later the patient died due to respiratory failure secondary to acute respiratory distress syndrome in shock state and acute kidney injury.
| Discussion | ▴Top |
AT/RTs are aggressive pediatric malignant brain tumors that accounts for approximately 1-2% of pediatric cancers of the CNS [5]. The majority of these tumors (approximately 60%) occur in the posterior cranial fossa (particularly the cerebellum) [6]. Only 45 cases of adults with AT/RTs have been reported in the literature to date [5]. Ostrom et al reported that the most common location in adults is the cerebral hemisphere, followed by the sellar region [7]. In contrast, Chan et al found that the most common location was the sellar region (46%), followed by cerebral hemisphere (32%) [8]. Sellar AT/RTs were reported to be almost exclusively in females and not in the pediatric population [9, 10]. Recently, it has been reported in the sellar region, with only 14 cases reported in the literature to date including our case (Table 1) [4, 6, 11-19]. All 14 cases are female of age range between 20 and 61 years old, and most of the cases presented with visual disturbance and ocular palsies.
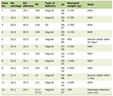 Click to view | Table 1. Summary of Previously Described Reports of Sellar AT/RTs and the Present Case |
Radiological findings of sellar AT/RTs are non-specific but are remarkably similar to pituitary adenoma; the lesions are isointense on T1-weighted imaging and enhance following gadolinium administration [12]. Histologically, AT/RTs are composed of diffuse proliferation of atypical large cells showing eccentrically located nuclei and abundant eosinophilic cytoplasm (rhabdoid features) with prominent nucleoli [4].
SMARCB1/INI1 is one of the core subunit proteins of the ATP-dependent SWI/SNF chromatin remodeling complex, and is identified as a potent and bona fide tumor suppressor [20]. Interactions have been demonstrated between SMARCB1/INI1 and key proteins in various pathways related to tumor proliferation and progression: the p16/RB pathway, WNT signaling3 pathway, sonic hedgehog signaling pathway and Polycomb pathway [20]. The molecular changes in AT/RTs include a mutation in one allele with a second allele loss due to monosomy 22, deletion of 22q11.2, or an acquired copy number neutral loss heterozygosity [21, 22]. Sellar AT/RTs also have a higher prevalence of biallelic INI1 alterations compared to AT/RTs in other locations [9]. Loss of expression of INI1 as detected by immunohistochemical staining correlates with deletion and mutations of the INI1 gene [23]. The differential diagnosis of sellar mass includes pituitary adenomas (85%), followed by craniopharyngioma (3%), Rathke cleft cyst (2%), and meningioma (1%) [24]. Pituitary adenoma shows uniform nuclear morphology with moderately abundant cytoplasm, and the neoplastic cells are highlighted by synaptophysin and chromogranin. Craniopharyngioma has a distinct morphology that the neoplastic cells are forming nodules or trabeculae of squamous epithelium, with peripheral nuclear palisading that surrounds looser plumper cells called “stellate reticulum”. In addition, nodules of anucleated squamous (“ghost” cells) and “wet” keratin are usually present. Rathke cleft cyst is a cyst lined by columnar ciliated epithelium with goblet cells. Meningioma usually has syncytial cells that have round uniform nuclei with intranuclear pseudo inclusions.
The prognosis of AT/RT is poor both in adults and in the pediatric population. Chan et al reported the average survival rate of adult AT/RT was 20 months [8]. This is comparable to the reported median survival of 13.5 to 16.8 months in the pediatric population [10, 25]. Long-term survival is possible in adult AT/RT cases after a combined approach including surgery, adjuvant radiotherapy, and chemotherapy [17, 26].
Conclusions
AT/RT is not limited to pediatric age group or to cerebellum, it should be considered in the differential diagnosis of malignant sellar lesion in adult patients. There is no specific radiological features that are available to differentiate AT/RT from pituitary adenoma, both of which represent as a sellar mass. Histologically, high grade features in form of necrosis, hemorrhage and frequent mitosis in adult sellar tumor should raise the possibility of ATRT. Use of BAF- 47(INI-1) immunohistochemical antibodies is the simple and quick tool to confirm the diagnosis. This high grade tumor with dismal outcome is surgically treated followed by radiochemotherapy.
Acknowledgments
None to declare.
Financial Disclosure
This study is not funded.
Conflict of Interest
The authors declare that they have no conflicts of interest.
Informed Consent
The patient died shortly after the surgery.
Author Contributions
Raghad Bokhari wrote the whole manuscript. Mohammed Bafaqeeh was the surgeon who did the surgery and provided the notes. Saad Al-Obaysi provided the radiology images with explanation for each image. Areej Al-Aman provided the clinical information. Wafa Alshakweer reviewed the manuscript.
| References | ▴Top |
- Biswas S, Wood M, Joshi A, Bown N, Strain L, Martinsson T, Campbell J, et al. Exome sequencing of an adult pituitary atypical teratoid rhabdoid tumor. Front Oncol. 2015;5:236.
doi pubmed - Rorke LB, Packer RJ, Biegel JA. Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg. 1996;85(1):56-65.
doi pubmed - Brada M. Pathology and genetics of tumours of the nervous system. Br J Cancer. 2001;84:148.
doi - Park HG, Yoon JH, Kim SH, Cho KH, Park HJ, Kim SH, Kim EH. Adult-onset sellar and suprasellar atypical teratoid rhabdoid tumor treated with a multimodal approach: a case report. Brain Tumor Res Treat. 2014;2(2):108-113.
doi pubmed - Souki C, Abdel-Rhaman M, Qasem A, Al-Hussaini M. Atypical teratoid rhabdoid tumor in adulthood. Clin Neuropathol. 2014;33(3):245-250.
doi pubmed - Almalki MH, Alrogi A, Al-Rabie A, Al-Dandan S, Altwairgi A, Orz Y. Atypical teratoid/rhabdoid tumor of the sellar region in an adult with long survival: case report and review of the literature. J Clin Med Res. 2017;9(3):216-220.
doi pubmed - Ostrom QT, Chen Y, P MdB, Ondracek A, Farah P, Gittleman H, Wolinsky Y, et al. The descriptive epidemiology of atypical teratoid/rhabdoid tumors in the United States, 2001-2010. Neuro Oncol. 2014;16(10):1392-1399.
doi pubmed - Chan V, Marro A, Findlay JM, Schmitt LM, Das S. A systematic review of atypical teratoid rhabdoid tumor in adults. Front Oncol. 2018;8:567.
doi pubmed - Nakata S, Nobusawa S, Hirose T, Ito S, Inoshita N, Ichi S, Amatya VJ, et al. Sellar Atypical Teratoid/Rhabdoid Tumor (AT/RT): A Clinicopathologically and Genetically Distinct Variant of AT/RT. Am J Surg Pathol. 2017;41(7):932-940.
doi pubmed - Nishikawa A, Ogiwara T, Nagm A, Sano K, Okada M, Chiba A, Agata M, et al. Atypical teratoid/rhabdoid tumor of the sellar region in adult women: Is it a sex-related disease? J Clin Neurosci. 2018;49:16-21.
doi pubmed - Kuge A, Kayama T, Tsuchiya D, Kawakami K, Saito S, Nakazato Y, Suzuki H. [Suprasellar primary malignant rhabdoid tumor in an adult: a case report]. No Shinkei Geka. 2000;28(4):351-358.
- Raisanen J, Biegel JA, Hatanpaa KJ, Judkins A, White CL, Perry A. Chromosome 22q deletions in atypical teratoid/rhabdoid tumors in adults. Brain Pathol. 2005;15(1):23-28.
doi pubmed - Arita K, Sugiyama K, Sano T, Oka H. Atypical teratoid/rhabdoid tumour in sella turcica in an adult. Acta Neurochir (Wien). 2008;150(5):491-495; discussion 496.
doi pubmed - Las Heras F, Pritzker KP. Adult variant of atypical teratoid/rhabdoid tumor: immunohistochemical and ultrastructural confirmation of a rare tumor in the sella tursica. Pathol Res Pract. 2010;206(11):788-791.
doi pubmed - Schneiderhan TM, Beseoglu K, Bergmann M, Neubauer U, Macht S, Hanggi D, Reifenberger G, et al. Sellar atypical teratoid/rhabdoid tumours in adults. Neuropathol Appl Neurobiol. 2011;37(3):326-329.
doi pubmed - Moretti C, Lupoi D, Spasaro F, Chioma L, Di Giacinto P, Colicchia M, Frajoli M, et al. Sella turcica atypical teratoid/rhabdoid tumor complicated with lung metastasis in an adult female. Clin Med Insights Case Rep. 2013;6:177-182.
doi pubmed - Chou SQH, Lo SSM, Wong HN, Lau P, Chan C, Tang K, Cheung YL. Atypical teratoid/rhabdoid tumour in the sella turcica of a female adult. Hong Kong J Radiol. 2013;16:65-68.
doi - Shitara S, Akiyama Y. Atypical teratoid/rhabdoid tumor in sellar turcica in an adult: A case report and review of the literature. Surg Neurol Int. 2014;5:75.
doi pubmed - Lev I, Fan X, Yu R. Sellar atypical teratoid/rhabdoid tumor: any preoperative diagnostic clues? AACE Clinical Case Reports. 2015;1(1):e2-e7.
doi - Kohashi K, Oda Y. Oncogenic roles of SMARCB1/INI1 and its deficient tumors. Cancer Sci. 2017;108(4):547-552.
doi pubmed - Biegel JA. Molecular genetics of atypical teratoid/rhabdoid tumors. Neurosurgical focus. 2006;20(1):1-7.
doi pubmed - Geller JI, Roth JJ, Biegel JA. Biology and treatment of rhabdoid tumor. Crit Rev Oncog. 2015;20(3-4):199-216.
doi pubmed - Judkins AR. Immunohistochemistry of INI1 expression: a new tool for old challenges in CNS and soft tissue pathology. Adv Anat Pathol. 2007;14(5):335-339.
doi pubmed - Goldblum JR, Lamps LW, McKenney JK, Myers JL. Rosai and Ackermans Surgical Pathology, Elsevier. 2017.
- Lafay-Cousin L, Hawkins C, Carret AS, Johnston D, Zelcer S, Wilson B, Jabado N, et al. Central nervous system atypical teratoid rhabdoid tumours: the Canadian Paediatric Brain Tumour Consortium experience. Eur J Cancer. 2012;48(3):353-359.
doi pubmed - Makuria AT, Rushing EJ, McGrail KM, Hartmann DP, Azumi N, Ozdemirli M. Atypical teratoid rhabdoid tumor (AT/RT) in adults: review of four cases. J Neurooncol. 2008;88(3):321-330.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Neurology Research is published by Elmer Press Inc.