

| Journal of Neurology Research, ISSN 1923-2845 print, 1923-2853 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Neurol Res and Elmer Press Inc |
| Journal website http://www.neurores.org |
Case Report
Volume 10, Number 1, February 2020, pages 20-24
Acute Reversible Cerebral Vasoconstriction Syndrome With Low-Dose Dihydroergotamine Possibly Potentiated by Valproic Acid and Erenumab: A Case Report
Hsiangkuo Yuana, Stephanie J. Nahasa, Matthew A. Berka, b
aDepartment of Neurology, Thomas Jefferson University Hospital, Philadelphia, PA 19107, USA
bCorresponding Author: Matthew A. Berk, Department of Neurology, Thomas Jefferson University, 909 Walnut Street, Philadelphia, PA 19107, USA
Manuscript submitted November 30, 2019, accepted December 6, 2019
Short title: RCVS With Low-Dose DHE
doi: https://doi.org/10.14740/jnr558
| Abstract | ▴Top |
Dihydroergotamine (DHE), in the setting of polypharmacy, may increase the possibility of reversible cerebral vasoconstriction syndrome (RCVS). A 64-year-old woman with chronic migraine and medication-overuse headache (on amitriptyline, duloxetine, erenumab) was electively admitted for 5 days of intravenous (IV) ketamine (up to 55 mg/h) to treat intractable migraine pain. On the seventh day, upon receiving her forth dose of IV DHE (0.25 mg) and the first of IV valproic acid (VPA) (500 mg) adjunctively, she developed acute bilateral decreased visual acuity, bitemporal visual field deficit, and unsteady gait. Brain magnetic resonance imaging/magnetic resonance angiography (MRI/MRA) showed confluent bilateral T2 hyperintensities with punctate restricted diffusion in the occipital lobes associated with multi-segmental narrowing involving anterior, middle, posterior cerebral, and basilar arteries consistent with RCVS. Verapamil was initiated, whereas DHE, neuroleptics, and serotonergic agents were discontinued. Though she continued to have constant, non-thunderclap migrainous headache, her other neurologic symptoms resolved in 24 h. Concomitant use of VPA and erenumab with DHE may result in RCVS. VPA likely displaces the protein-bound DHE causing a transient surge of free DHE level in the serum. Erenumab may have impaired the protective vasodilatory mechanism, augmenting DHE’s vasoconstrictive effect. This case report highlights the importance and awareness of such a drug-drug interaction with DHE.
Keywords: Reversible cerebral vasoconstriction syndrome; Posterior reversible encephalopathy syndrome; Valproate; Valproic acid; Dihydroergotamine; Migraine; Erenumab
| Introduction | ▴Top |
Clinical management of migraine, a disease characterized by disabling headache attacks believed to be due to trigeminovascular dysfunction, can be challenging. Migraine preventives carried over from medications designed for other medical conditions (e.g., antiepileptics, antihypertensives, antidepressants) commonly display unique adverse event (AE) profiles that can lead to reduced medication adherence and poor migraine control [1]. In refractory migraine, use of intravenous (IV) medications such as lidocaine, dihydroergotamine (DHE), or ketamine may offer acute relief [2, 3]. Despite achieving greater headache control by using a cocktail of medications, unexpected AEs may occur, especially in the setting of polypharmacy. Here, we report a case of a woman who developed posterior reversible encephalopathy syndrome (PRES) and reversible cerebral vasoconstriction syndrome (RCVS) while being treated with IV ketamine, neuroleptics, DHE, and other medications in a hospital setting.
DHE, an ergot derivative synthesized in the 1940s, interacts with multiple serotoninergic receptors blocking the release of neuropeptides and subsequent neurogenic inflammation and vasodilation in migraine attacks [4]. It is commonly believed that DHE produces sustained migraine relief via 5-hydroxytryptamine (5HT) 1B/1D agonism with longer and stronger receptor binding than sumatriptan [5]. The therapeutic effect of DHE does not correlate with its serum concentration, probably due to the longer half-life of the active metabolites [6]. Although both IV and intranasal DHE have superior efficacy compared to placebo in randomized clinical trials [7, 8], DHE’s clinical utility can be hampered by unwanted AEs, including nausea/vomiting and vasoconstriction, particularly when used in the presence of potent enzyme inhibitors (e.g., macrolides, certain antivirals, and antifungals). DHE reduces carotid arteriovenous anastomosis via the 5HT1B receptor and α2A/2C adrenoreceptor agonism in a dose-dependent fashion [9]. DHE and its metabolites (8’-OH-DHE and 8’,10’-OH DHE) also reduce venous compliance [6], and it is a more potent vasoconstrictor than meningeal/coronary arterial vasoconstrictor [10, 11]. Here, we report a previously DHE-tolerant patient developing DHE-triggered vasospasm possibly facilitated by valproic acid and erenumab.
| Case Report | ▴Top |
A 64-year-old woman with a history of chronic migraine and medication-overuse headache was admitted for management of intractable migraine. She had episodic migraine in her 20s and 30s followed by complete and seemingly spontaneous remission until after menopause, when her migraine returned and eventually became continuous and debilitating. She experienced acute and preventive treatment failures from non-steroidal anti-inflammatory drugs, triptans, topiramate, propranolol, amitriptyline, onabotulinumtoxinA, sphenopalatine ganglion block, greater occipital nerve block, medical marijuana, transcutaneous supraorbital nerve stimulation, and many other agents. Upon presentation to our institution, she was taking oral divalproex sodium and oral morphine. She underwent two hospitalizations in 2017 wherein she responded well to lidocaine infusion and successfully weaned off of morphine each time. However, she experienced relapse within weeks of discharge, leading to her recurrent reliance on opioids. During each admission, she tolerated maximal doses of DHE (1 mg every 8 h), various neuroleptics, magnesium sulfate, ketorolac, and methylprednisolone without significant AEs. Upon discharge, she was given a prescription for ketamine nasal spray to try on an introductory basis as an outpatient. She subsequently required cholecystectomy, which was believed due to ketamine nasal spray overuse. Despite using multiple migraine preventives (duloxetine, amitriptyline, erenumab (started June 2018 at 70 mg, increased to 140 mg in August 2018), single pulse transcranial magnetic stimulation), she remained dependent on morphine (36 mg/day) with daily headache (intensity 3 - 10/10, average 7/10) prior to this admission. In mid-September 2018, she was admitted electively for ketamine infusion and detoxification from morphine.
Other pertinent history includes irritable bowel syndrome with constipation, hypothyroidism, remote focal subarachnoid hemorrhage of unclear etiology, anxiety/depression, remote smoking (quit 19 years prior), and no ethanol abuse.
On admission exam, she was well nourished (body mass index (BMI) 22.7 kg/m2) with normal vital signs, normal general exam, and no focal neurological deficits. Complete metabolic panel and blood count were unremarkable.
Figure 1 details her medication regimen during her hospitalization. Upon admission, she was placed on a standard migraine IV cocktail (magnesium sulfate, ketorolac, neuroleptic) along with a ketamine infusion, which was carefully titrated up to 55 mg/h (0.89 mg/kg/h) while monitoring for AEs (e.g., hypertension, hallucination and anxiety). A clonidine patch was used routinely to counteract ketamine-associated sympathomimetic AEs, including hypertension. DHE 0.5 mg was added on the third day for two doses but was stopped due to chest pain. It was restarted on the sixth day at 0.25 mg every 8 h (q8h) along with methadone (for morphine detoxification) and methylprednisolone (replacing ketorolac) as her headache remained poorly controlled. Ketamine was tapered down starting on day 5 and discontinued on day 6. On the seventh day, her headache remained a 9/10. She received her last IV DHE 0.25 mg at 11:52 am, her first chlorpromazine 6.25 mg (replacing promethazine) IV dose at 12 pm, and first valproic acid (VPA) 500 mg IV dose at 12:26 pm that day.
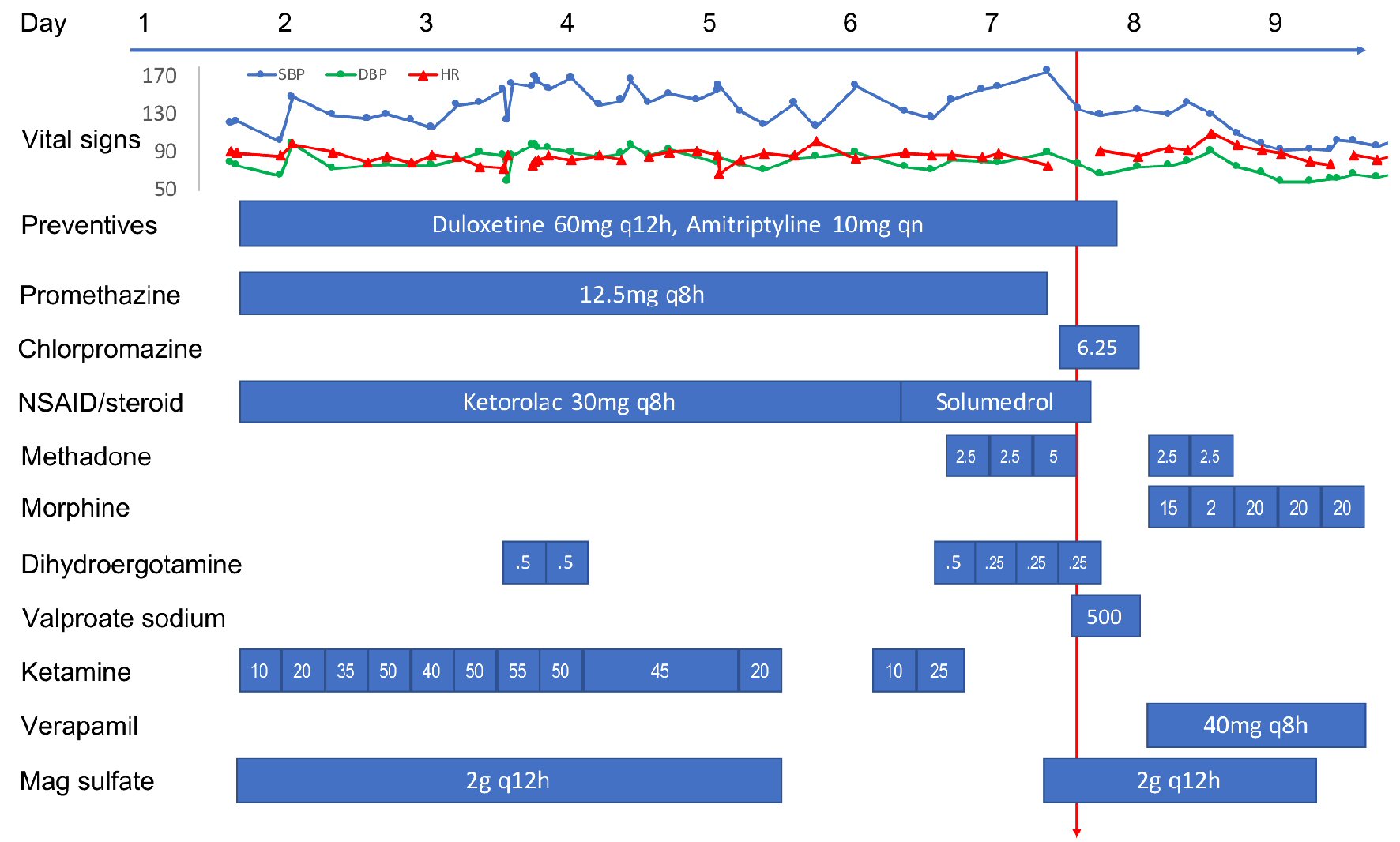 Click for large image | Figure 1. Time course of vital signs and medications administered to the patient during the hospitalization. All medications were intravenous formulation except oral preventives, methadone/morphine, and verapamil. All doses are in mg except where noted. Red arrow indicates the time of PRES/RCVS onset. PRES: posterior reversible encephalopathy syndrome; RCVS: reversible cerebral vasoconstriction syndrome; NSAID: non-steroidal anti-inflammatory drug. |
Around 1 - 1:30 pm, while still experiencing severe headache, she developed acute onset of visual change, initially described as double vision and later as blurry, as if “looking through a veil.” On exam, she had normal vital signs and mentation, but displayed reduced visual acuity (worse on the left) with no afferent pupillary defect. Visual field deficit was difficult to ascertain accurately but appeared to be bitemporal. Other cranial nerves, motor/sensory, coordination and tendon reflex exams were normal, except for mild unsteady gait due to subjective dizziness. DHE, chlorpromazine, VPA, methylprednisolone, amitriptyline, and duloxetine were all discontinued. Around 2 pm, her visual acuity and visual fields seemed to improve, but she declined complete ophthalmologic reassessment due to headache and photosensitivity. Brain computed tomography (CT) in the afternoon found bilateral occipital low attenuation. Brain magnetic resonance imaging (MRI) performed later at night showed abnormal T2 hyperintensity in bilateral parietal, occipital, and posterior temporal white matter with small areas of restricted diffusion in the bilateral occipital lobes (Fig. 2). Brain magnetic resonance angiography (MRA) revealed diffuse multi-segmental narrowing involving the bilateral anterior cerebral, middle cerebral, posterior cerebral and basilar arteries (Fig. 2). The findings were deemed consistent with PRES and RCVS. On the next day, her visual and gait deficits completely resolved. She was placed on verapamil and morphine for 3 days and then discharged home. She has lost follow-up ever since. At the time of writing, she is having only a few migraines a month managed by topiramate, fremanezumab, and cognitive behavioral therapy.
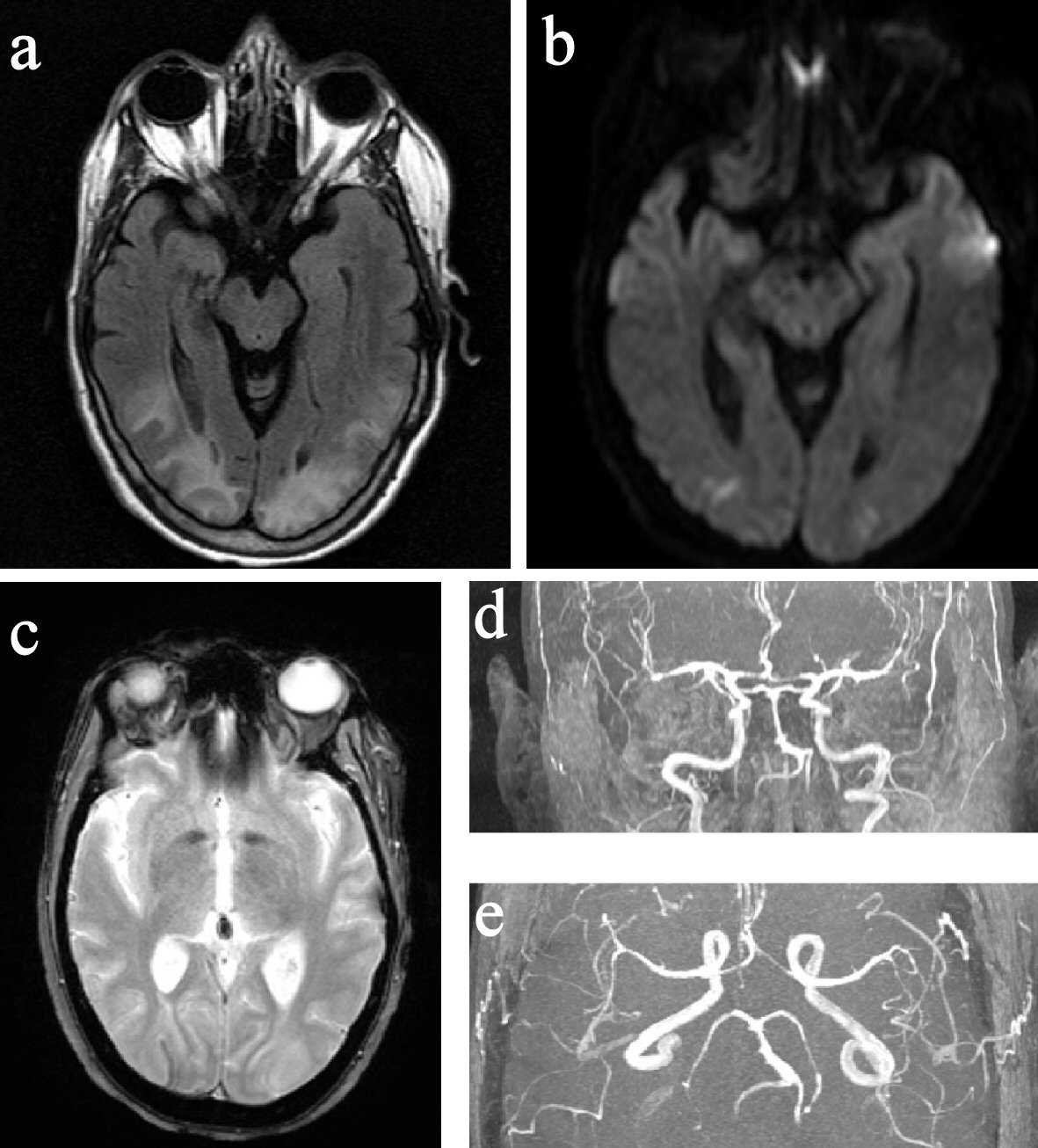 Click for large image | Figure 2. MRI and MRA of the brain. (a) FLAIR image showed hyperintensity in the posterior parietal and occipital lobes. (b) DWI image showed small bilateral occipital pole diffusion restrictions. (c) GRE image showed right occipital curvilinear superficial siderosis. (d) and (e) Segmental narrowing of bilateral ACA, MCA, PCA, and BA. MRI: magnetic resonance imaging; MRA: magnetic resonance angiography; FLAIR: fluid-attenuated inversion recovery; DWI: diffusion-weighted imaging; GRE: gradient-recalled echo; ACA: anterior cerebral artery; MCA: middle cerebral artery; PCA: posterior cerebral artery; BA: basilar artery. |
| Discussion | ▴Top |
Based on the clinical course, her RCVS occurred after receiving chlorpromazine, DHE, and VPA while on magnesium sulfate, duloxetine, amitriptyline, erenumab, methylprednisolone, and methadone. Ketamine infusion had been discontinued the night before. Reviewing the AE profiles from these medications, the most plausible causative agent for PRES/RCVS is DHE, but with some caveats. Since our patient was tolerant to maximal doses of DHE (1 mg q8h) in the past, we propose and discuss four possible explanations for vasospasm under lower-dose DHE (0.25 mg q8h).
The first is the presence of enzyme inhibitors that increase the DHE serum level. DHE is primarily metabolized by cytochrome P450 3A4 (CYP3A4) in the liver with an α phase half-life of 1.45 h and β phase half-life of 15 h [12]. We reviewed all the medications with possible enzyme inhibition activity (Table 1); none were strongly affecting CYP3A4. So, this is likely not the main reason.
The second is the presence of other vasoactive agents that potentiates the DHE effect. Ketamine is a noncompetitive N-methyl-D-aspartate receptor antagonist that also affects sodium- and voltage-dependent calcium channels, and activates nitric oxide cyclic guanosine monophosphate pathways [13]. These properties likely have no role in vasoconstriction. In addition, ketamine and duloxetine may increase sympathetic activity as both are associated with hypertension. However, in our case, the patient’s blood pressures were within normal range (with the use of clonidine patch) during the entire hospitalization. Furthermore, ketamine had also been discontinued for more than 12 h when PRES/RCVS occurred. So, with ketamine’s half-life of 2 - 4 h, it is likely not the main contributor.
The third possible explanation is that the release of free DHE from protein-bound DHE creates a transient level surge. DHE distributes very quickly, with only 6% of intravenously administered doses observed in plasma in 5 min. DHE not only binds highly to plasma proteins (94-99%), but also distributes highly to blood cells (> 50%) [12]. Temporally, the visual symptoms started 30 - 60 min after VPA/chlorpromazine infusion. VPA, which is also highly protein bound, can compete for protein binding, resulting in displacement of DHE. Such a phenomenon has been observed from VPA on phenytoin [14]. Chlorpromazine can also displace DHE, but it likely plays a less significant role given its lower dosing (6.25 mg) compared to VPA (500 mg). This mechanism seems a plausible explanation for potentiating the DHE vasoconstriction and PRES/RCVS.
The forth one is the calcitonin-gene related peptide (CGRP) functional blockade hampers CGRP’s protective vasodilatory effect: erenumab blocks CGRP receptors. Given that CGRP is a potent vasodilator, blockade of its activity particularly at the receptor level could theoretically lead to derangements in cerebrovascular tone autoregulation [15]. It is worth noting that CGRP receptor blockade reverses CGRP-induced vasodilation, but does not cause vasoconstriction in cranial arteries [16]. Therefore, erenumab may hamper CGRP’s protective vasodilatory effect thereby indirectly augmenting DHE’s vasoconstrictive AE [17].
This case report is limited by a lack of a re-challenge test. Based on the Naranjo adverse drug reaction scale, we can only assume a probable correlation. Since re-challenging is clearly unethical, more clinical awareness about this potential drug interaction with DHE would help in identifying similar cases and avoiding the same outcome.
Conclusions
We report a rare but serious complication (PRES/RCVS) of DHE in the setting of polypharmacy. A clear response to a lower dose of DHE prompted us to look for DHE augmenting factors. The infusion of VPA likely displaced the protein-bound DHE causing a transient surge of free DHE in the serum that subsequently potentiated the vasoconstrictive effect. While VPA is commonly known for its enzyme inhibitory effect, the displacement of protein-bound medication is a documented phenomenon but less well recognized. In addition, treatment with erenumab may have impaired the protective vasodilatory mechanisms for cerebrovascular autoregulation from CGRP. Lack of such protective vasodilation may inadvertently augment DHE’s vasoconstrictive effect. This case report highlights the importance of awareness of potential drug-drug interactions with DHE.
Acknowledgments
The authors would like to thank Anna Chen for her careful proofreading.
Financial Disclosure
None to declare.
Conflict of Interest
HY has received honoraria from Supernus Pharmaceutical. SN has received personal compensation for authorship/editing from Springer and Woulters-Klouwer and for consulting/speaking from Allergan, Amgen/Novartis, Biohaven, electroCore, Ely Lilly, Supernus, Theranica, and Teva. MB has nothing to disclose.
Informed Consent
A written informed consent was obtained from the patient.
Author Contributions
HY, SN, MB conceived and designed the manuscript. HY drafted the manuscript. SN and MB performed critical revisions and approved the final version.
| References | ▴Top |
- Hepp Z, Dodick DW, Varon SF, Gillard P, Hansen RN, Devine EB. Adherence to oral migraine-preventive medications among patients with chronic migraine. Cephalalgia. 2015;35(6):478-488.
doi pubmed - Pomeroy JL, Marmura MJ, Nahas SJ, Viscusi ER. Ketamine Infusions for Treatment Refractory Headache. Headache. 2017;57(2):276-282.
doi pubmed - Rosen N, Marmura M, Abbas M, Silberstein S. Intravenous lidocaine in the treatment of refractory headache: a retrospective case series. Headache. 2009;49(2):286-291.
doi pubmed - Silberstein SD, McCrory DC. Ergotamine and dihydroergotamine: history, pharmacology, and efficacy. Headache. 2003;43(2):144-166.
doi pubmed - Kori S, Zhang J, Kellerman D, Armer T, Goadsby P. Sustained pain relief with dihydroergotamine in migraine is potentially due to persistent binding to 5-HT1B and 5-HT1D receptors. J Headache Pain. 2013;14(S1):507.
doi - Muller-Schweinitzer E. Pharmacological actions of the main metabolites of dihydroergotamine. Eur J Clin Pharmacol. 1984;26(6):699-705.
doi pubmed - Gallagher RM. Acute treatment of migraine with dihydroergotamine nasal spray. Dihydroergotamine Working Group. Arch Neurol. 1996;53(12):1285-1291.
doi pubmed - Callaham M, Raskin N. A controlled study of dihydroergotamine in the treatment of acute migraine headache. Headache. 1986;26(4):168-171.
doi pubmed - Villalon CM, Centurion D, Willems EW, Arulmani U, Saxena PR, Valdivia LF. 5-HT1B receptors and alpha 2A/2C-adrenoceptors mediate external carotid vasoconstriction to dihydroergotamine. Eur J Pharmacol. 2004;484(2-3):287-290.
doi pubmed - de Hoon JN, Poppe KA, Thijssen HH, Struijker-Boudier HA, Van Bortel LM. Dihydroergotamine: discrepancy between arterial, arteriolar and pharmacokinetic data. Br J Clin Pharmacol. 2001;52(1):45-51.
doi pubmed - Labruijere S, Chan KY, de Vries R, van den Bogaerdt AJ, Dirven CM, Danser AJ, Kori SH, et al. Dihydroergotamine and sumatriptan in isolated human coronary artery, middle meningeal artery and saphenous vein. Cephalalgia. 2015;35(2):182-189.
doi pubmed - Wyss PA, Rosenthaler J, Nuesch E, Aellig WH. Pharmacokinetic investigation of oral and i.v. dihydroergotamine in healthy subjects. Eur J Clin Pharmacol. 1991;41(6):597-602.
doi pubmed - Sleigh J, Harvey M, Voss L, Denny B. Ketamine - More mechanisms of action than just NMDA blockade. Trends in Anaesthesia and Critical Care. 2014;4(2-3):76-81.
doi - Dahlqvist R, Borga O, Rane A, Walsh Z, Sjoqvist F. Decreased plasma protein binding of phenytoin in patients on valproic acid. Br J Clin Pharmacol. 1979;8(6):547-552.
doi pubmed - Hong KW, Pyo KM, Lee WS, Yu SS, Rhim BY. Pharmacological evidence that calcitonin gene-related peptide is implicated in cerebral autoregulation. Am J Physiol. 1994;266(1 Pt 2):H11-16.
doi pubmed - Edvinsson L, Chan KY, Eftekhari S, Nilsson E, de Vries R, Saveland H, Dirven CM, et al. Effect of the calcitonin gene-related peptide (CGRP) receptor antagonist telcagepant in human cranial arteries. Cephalalgia. 2010;30(10):1233-1240.
doi pubmed - Grell AS, Haanes KA, Johansson SE, Edvinsson L, Sams A. Fremanezumab inhibits vasodilatory effects of CGRP and capsaicin in rat cerebral artery - Potential role in conditions of severe vasoconstriction. Eur J Pharmacol. 2019;864:172726.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Neurology Research is published by Elmer Press Inc.