

| Journal of Neurology Research, ISSN 1923-2845 print, 1923-2853 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Neurol Res and Elmer Press Inc |
| Journal website https://www.neurores.org |
Case Report
Volume 10, Number 6, December 2020, pages 240-244
Diffuse Astrocytoma and Ollier’s Disease
Assem S. Al Rumeha, e, Toleen Shaikhb, Ahmad Larib, Abdullah A. Muharibc, Wafaa Al Shakweerd
aPathology and Clinical Laboratory Medicine Administration, Prince Sultan Military Medical City, Riyadh, Saudi Arabia
bSaudi Boared Emergency Medicine, Royal Commission Hospital, Al Jubail, Saudi Arabia
cDepartment of Neurosurgery, King Fahad Medical City, Riyadh, Saudi Arabia
dDepartment of Radiology, King Fahad Medical City, Riyadh, Saudi Arabia
eCorresponding Author: Assem S. Al Rumeh, Pathology and Clinical Laboratory Medicine Administration, Prince Sultan Military Medical City, Riyadh, Saudi Arabia
Manuscript submitted March 30, 2020, accepted July 7, 2020, published online December 9, 2020
Short title: Diffuse Astrocytoma and Ollier’s Disease
doi: https://doi.org/10.14740/jnr580
| Abstract | ▴Top |
Several syndromes are associated with an increased incidence of intracranial tumors. Among these is enchondromatosis. It is a rare, nonhereditary condition. The common subtypes of enchondromatosis are Ollier’s disease (OD) and Maffucci syndrome. It has been distinguished between them by the presence of vascular malformation and non-skeletal neoplasm in Maffucci syndrome. The emergence of malignant neoplasms, including gliomas is a well-recognized complication in Maffucci syndrome, but over the past few years there has been an increasing number of reported cases of patients with intracranial tumors and OD. In our case report we discuss a 23-year-old female recently diagnosed with World Health Organization (WHO) grade II diffuse astrocytoma and found to have OD.
Keywords: Ollier’s disease; Maffucci syndrome; Gliomas; Diffuse astrocytoma; Dysembrionic neuroepithelial tumors; Gangliogliomas
| Introduction | ▴Top |
Malignant gliomas are tumors of glial origin of the central nervous system (CNS), exhibiting various degrees of differentiation [1]. Several syndromes are associated with an increased incidence of intracranial tumors. Among these is enchondromatosis. Enchondromas are common benign usually asymptomatic cartilaginous tumors [2, 3]. Enchondromatosis or Ollier’s disease (OD, World Health Organization (WHO) terminology) [2] is defined by the presence multiple enchondromas and characterized by an asymmetric distribution of cartilaginous lesions that can be extremely variable. It is first described by Maffucci in 1881 in association with venous angiomas [4]. In 1899, Ollier described enchondromatosis in a patient with no evidence of vascular anomalies, known as OD [4]. The emergence of malignant neoplasms including gliomas is a well-recognized complication in Maffucci syndrome [2, 3, 5].
| Case Report | ▴Top |
A 23-year-old female presented at the clinic with 2 months’ history of frontal mild pressure like headache that was managed by over the counter analgesia, and two episodes of mal seizure during sleep that last for 15 min. Neurological examination at time of presentation was normal. Upon physical examination, abnormal boney lesions were noticed in the left shoulder and left iliac bone.
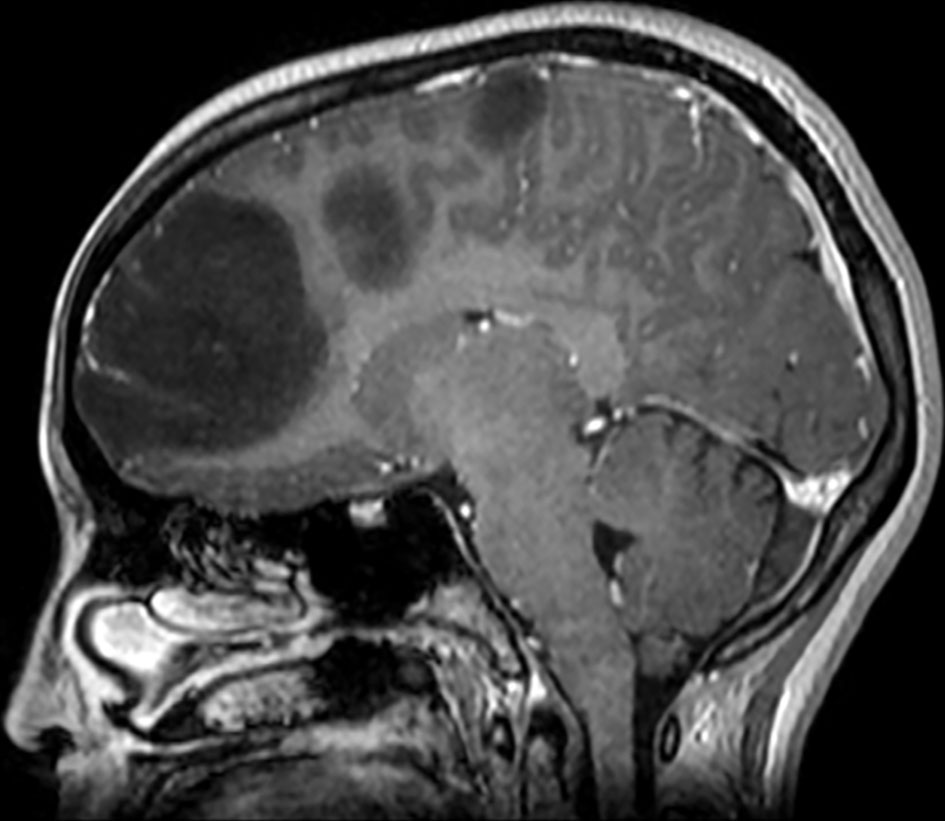
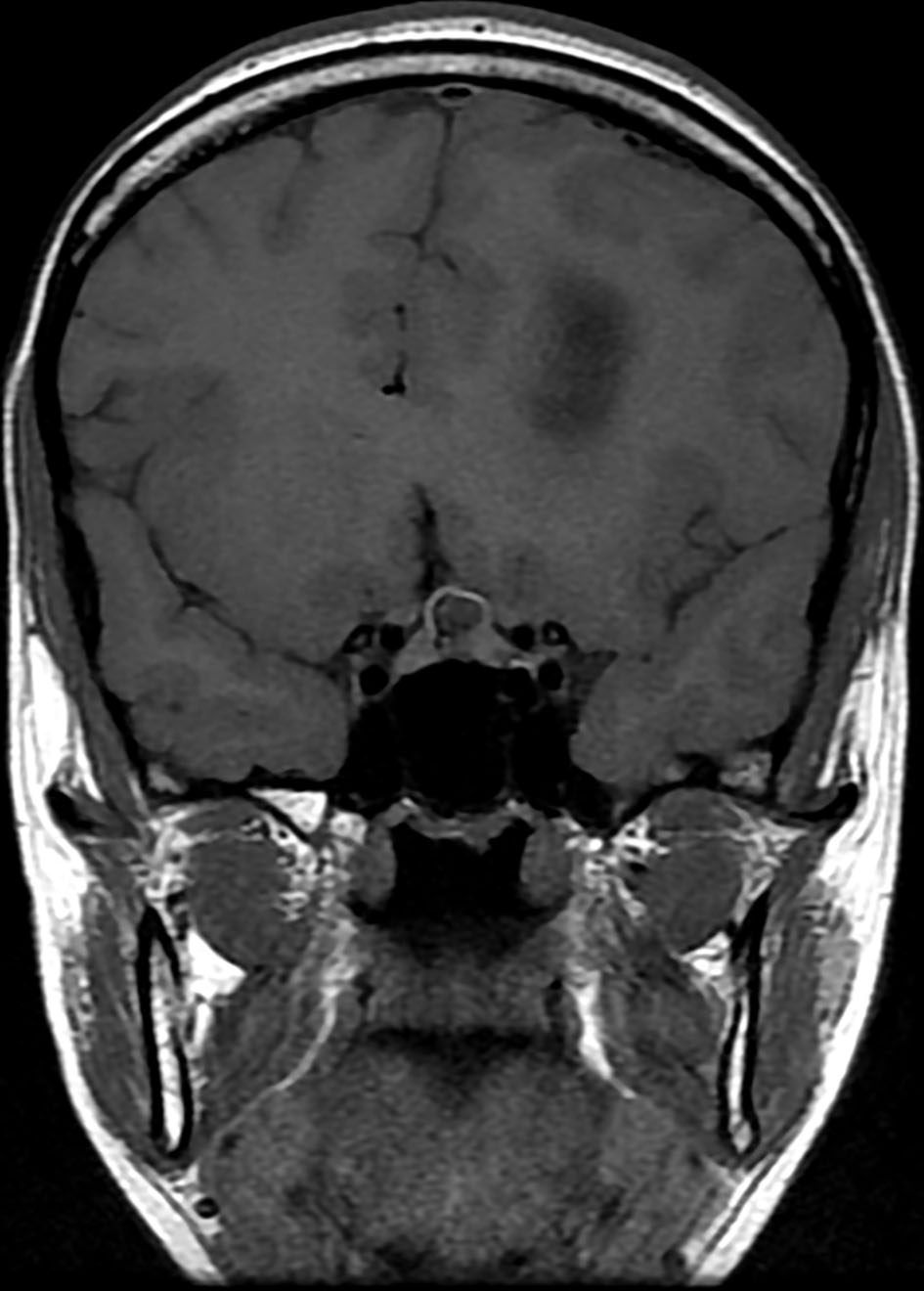
Brain magnetic resonance imaging (MRI) showed three left frontal cortical based lesions which appeared hypointense on T1 weighted image (T1WI) and hyperintense on T2WI with mild mass effect without significant vasogenic edema. The largest lesion measured 5.6 × 5.4 × 4.3 cm, also showed inhomogeneity with cystic changes and modest enhancement after intravenous (IV) gadolinium administration. Also present was a tiny ring enhancing focus exhibiting restricted diffusion. The other two lesions had no enhancement nor diffusion restriction (Figs. 1, 2). In addition, a small complex sellar lesion with anterior enhancing component was demonstrated, it was extending into the suprasellar cistern and abutting the optic chiasm and the pre-chiasmatic portions of the optic nerves. Also noted was a cerebellar tonsillar ectopia (Fig. 3).
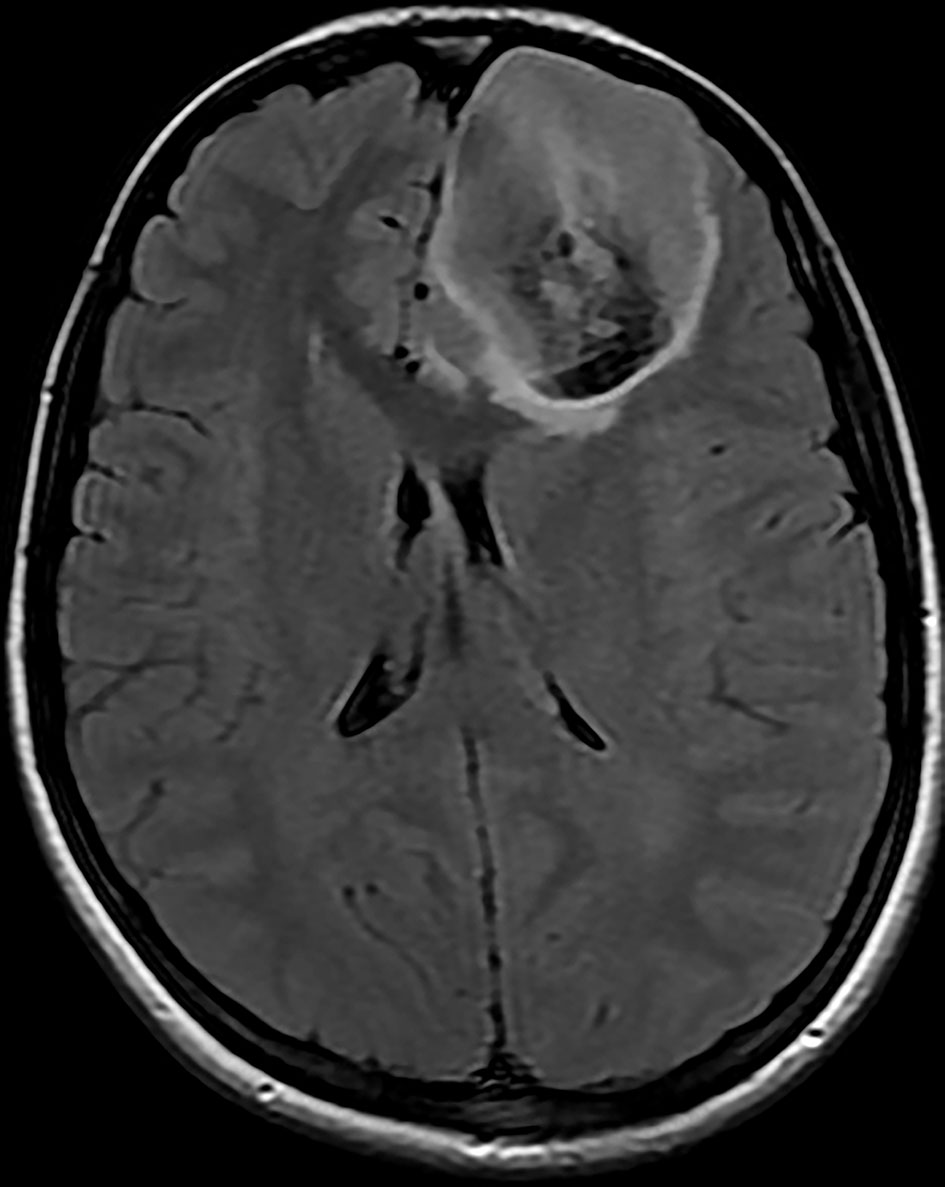 Click for large image | Figure 1. Brain MRI shows three left frontal cortical based lesions which appear hyperintense on T2 weighted image with mild mass effect but without significant vasogenic edema. The largest lesion shows inhomogeneity with cystic changes and modest enhancement after IV gadolinium administration with tiny ring enhancing focus which shows restricted diffusion. MRI: magnetic resonance imaging; IV: intravenous. |
 Click for large image | Figure 2. Brain MRI shows three left frontal cortical based lesions which appear hypointense on T1 weighted image. MRI: magnetic resonance imaging. |
 Click for large image | Figure 3. Brain MRI shows a small complex sellar lesion with anterior enhancing component. It is extending into the suprasellar cistern. MRI: magnetic resonance imaging. |
Computed tomography (CT) brain without contrast of the skull base demonstrated calcified small seller/suprasellar lesion, Chiari malformation type I and asymmetrical occipital condyles. The left enlarged condyle was radiolucent compared to the normal right condyle. She underwent left frontal craniotomy with the neuro-navigation guidance.
Hematoxylin and eosin-stained sections from the 2 × 2 cm soft and pinkish neuro- specimen showed infiltrative low-grade glioma with mixed morphology and microcytic foci. It was composed of uniform round to oval cells with fried egg appearance. There was neither evidence of mitotic activity, endothelial vascular proliferation nor necrosis (Figs. 4, 5). Immunohistochemical study was performed on paraffin embedded tissue revealed negative reaction with p53 antibody. Isocitrate dehydrogenase 1 (IDH1, mutantR132H) stain was negative too (Figs. 6, 7). IDH1/2 pyrosequencing was showed positive result as IDH1 codon 132 c.394C>T p. Arg132Cys (R132C) mutation was also detected. Cytogenetic study of 1p19q revealed intact 1p36 19q13 which confirmed diffuse astrocytoma, WHO grade II diagnosis.
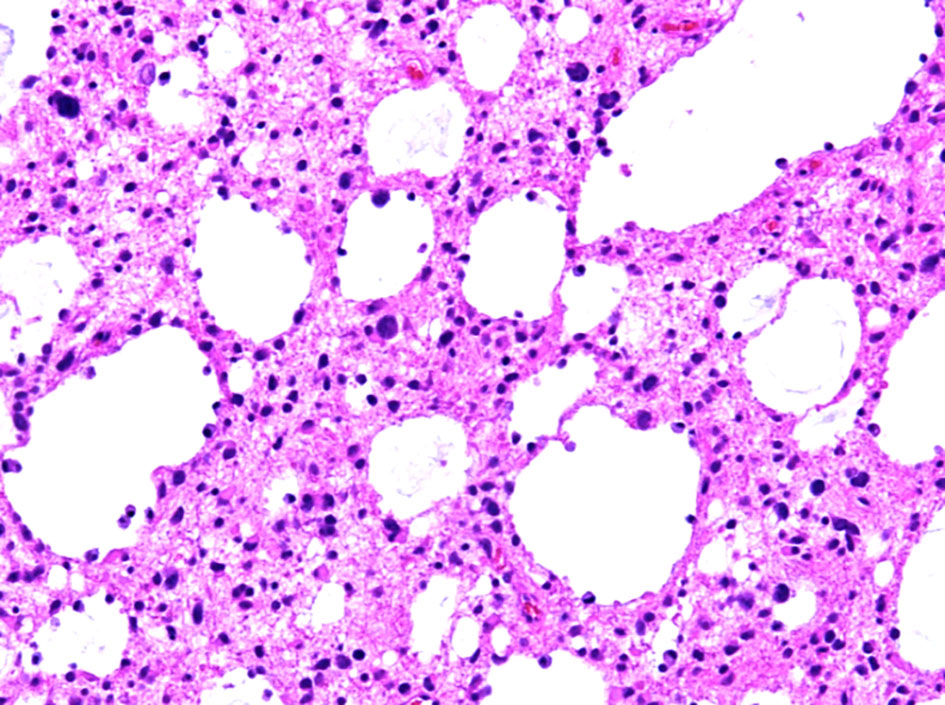 Click for large image | Figure 4. Histopathological examination of the diffuse astrocytoma specimen at intermediate power (× 20) shows an infiltrative growth pattern and microcytic changes. |
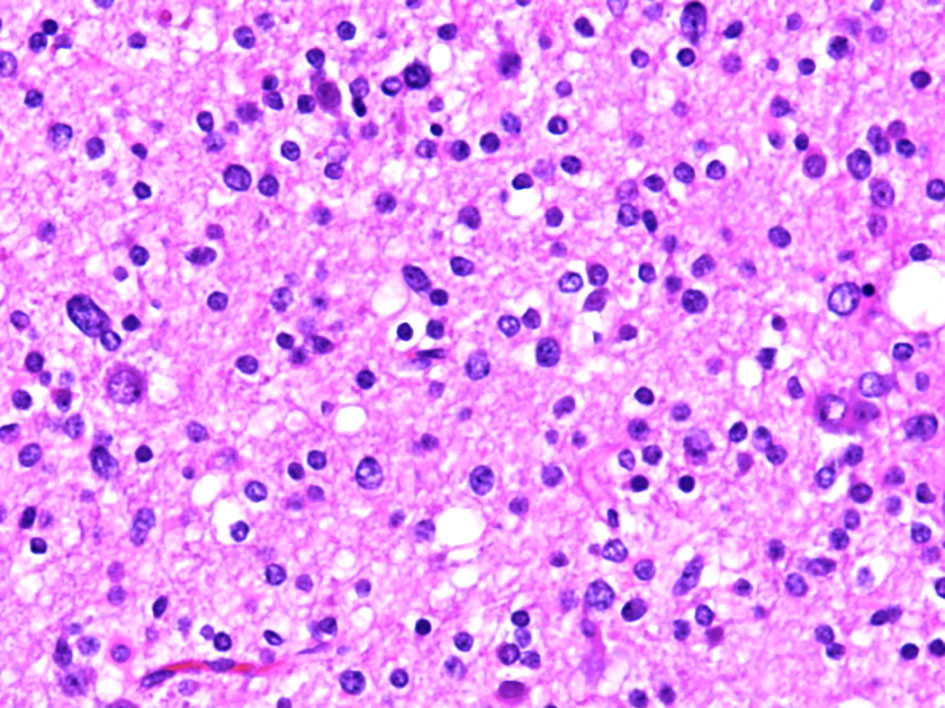 Click for large image | Figure 5. Histopathological examination of the diffuse astrocytoma specimen at high power (× 40) show a tumor cells have a more round and uniform appearance with microcytic changes. |
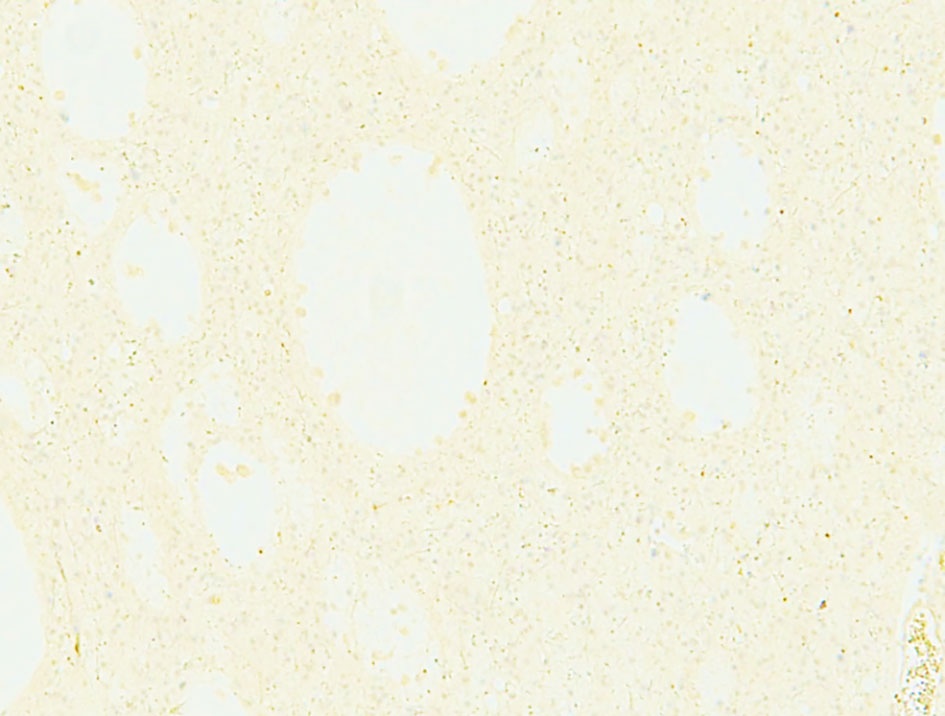 Click for large image | Figure 6. Negative immunohistochemical staining for IDH1. IDH1: isocitrate dehydrogenase 1. |
 Click for large image | Figure 7. Ki67 immunohistochemical staining shows from low to focally moderate (up to 6-8%). |
X-ray of the pelvis showed ill-defined mixed lytic and sclerotic lesions with chondroid matrix irregularity and bony destruction affecting mainly the left iliac bone, to a lesser extent the left inferior pubic ramus and the iliac side of the right sacroiliac (SI) joint. No periosteal reaction nor pathological fracture was present.
Ill-defined mixed lytic and sclerotic lesion at the proximal metadiaphyseal region of the left humerus was also demonstrated, causing cortical distraction. Lamellated and Codman periosteal reaction were noted as well as a wide zone of transition. Dorsal and radial bowing of the radius with exaggerated radial inoculation, dislocation of the proximal radial head, shortening and bowing of the ulna were all suggesting Madelung’s deformity. Proximal fourth and fifth phalanges also showed lytic lesions. These features were suggestive of polyostotic enchondromatosis (OD) (Fig. 8).
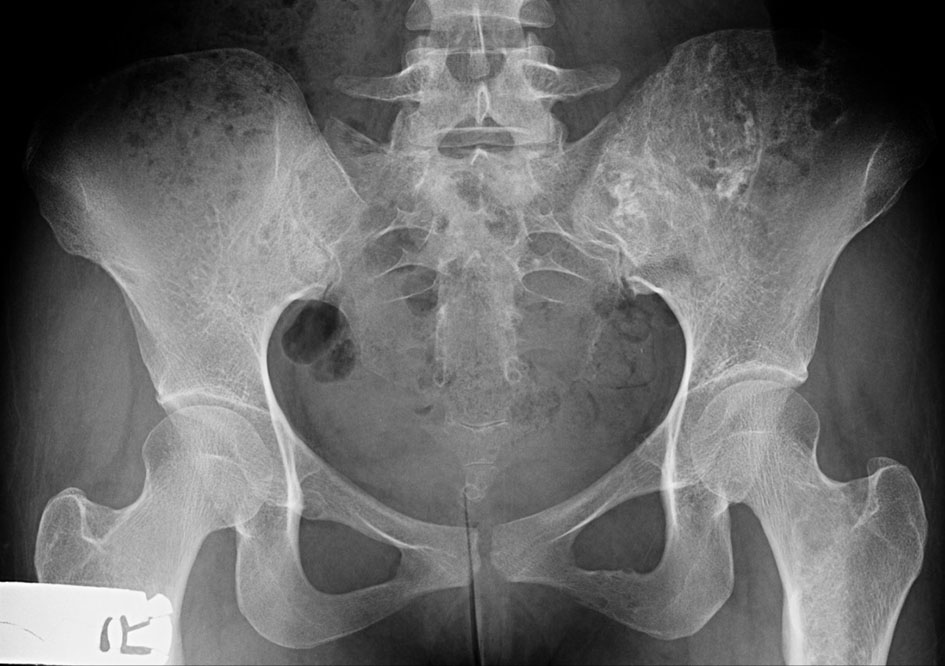 Click for large image | Figure 8. X-ray of the pelvis (AP view) shows ill-defined mixed lytic and sclerotic lesions with chondroid matrix, irregularity and bony destruction affecting mainly the left iliac bone and to a lesser extent the left inferior pubic ramus and the iliac side of the right SI joint. No periosteal reaction nor pathological fracture. AP: anteroposterior; SI: sacroiliac. |
Enhanced chest CT showed left upper lobe few nonspecific subpleural nodules. There was an intraluminal filling defect in the inferior division of the right lower lobe pulmonary artery. Also, there were multiple osseous expansile lesions with popcorn type calcification. No evidence of metastasis (Fig. 9).
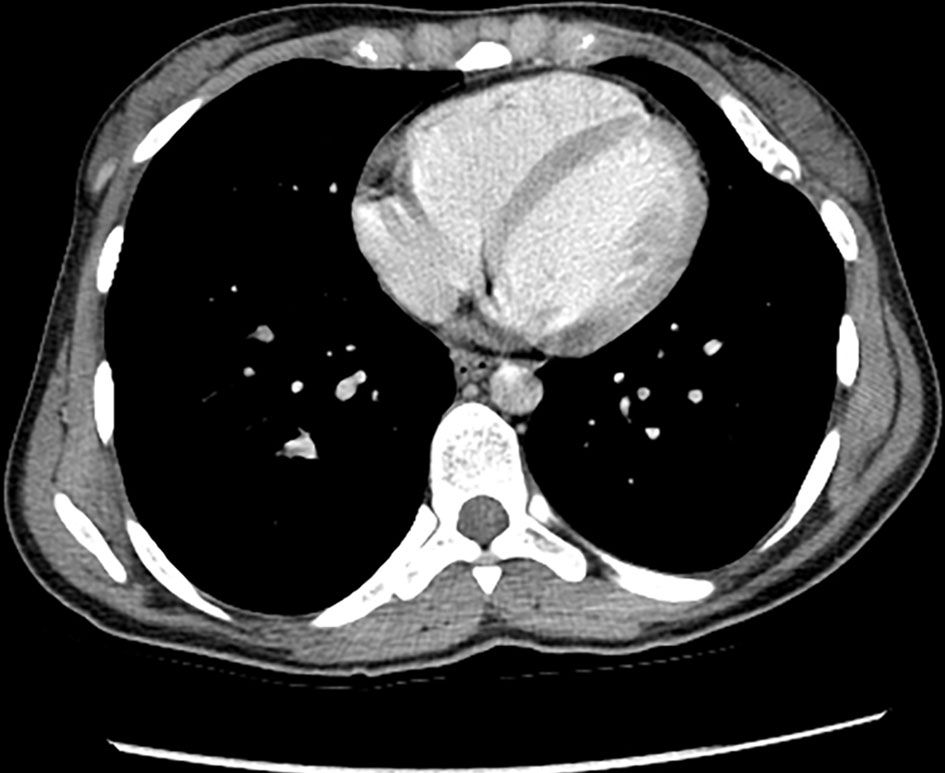 Click for large image | Figure 9. Enhanced chest CT (axial view) shows few nonspecific subpleural nodules in the left upper lobe. There are multiple osseous expansile lesions with popcorn type calcification. CT: computed tomography. |
CT scan-guided biopsy of the left iliac bone lesion confirmed the benign cartilaginous proliferation, consistent with chondroma. It is negative for malignancy and inflammation. Patient was discharged and was followed up with oncology department and genetics.
| Discussion | ▴Top |
There have been 25 reported cases in the literature including our patient since 1979 (Table 1) [4, 6-9].
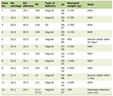 Click to view | Table 1. Twenty-Five Reported Cases in the Literature Including Our Patient Since 1979 |
A retrospective analysis of these cases showed the age of the patients ranged from four to 55. The majority were males in their second decade of life. This is similar to the observations of Bathla et al and Hori et al [4, 8].
Nine of the 25 cases were of a high grade. One case was reported as astrocytoma with no further grading. There was one reported case of oligodendroglioma and four cases with oligoastrocytoma [4]. Our case is representing the twelfth low grade astrocytoma associated with OD [4, 7].
Over the past few years OD patients seem to have an increased risk for the development of non-skeletal malignancies, especially intracranial tumors of glial origin [6, 9]. Our patient along with the previously reported cases points to the association between OD and glial tumors. Our patient along with the previously reported cases points to the association between OD and glial tumors.
Actually, the morphology of the brain tumor in our case was that of oligodendroglioma with infiltrative nature, microcysts formation and fried egg appearance. However, the immunohistochemical study for IDH1 (mutant R132H) was negative, in such cases and according to the WHO of the CNS tumors 2016, it should be cytogenetically studied for both IDH-1/2 and 1p19q to reach to the definitive diagnosis [10].
1p19q codeletion is an important diagnostic cytogenetic tool differentiating diffuse astrocytoma from oligodendroglioma, 1p19q is normal in the former and codeleted in oligodendroglioma regardless of the grade. This rule is only applied to adult oligodendroglioma. Oligodendroglioma of adult patients exhibit mutation of IDH1/2, 1p36 and 19q13 codeletion, and intact ATRX while the pediatrics’ oligodendroglioma have normal 1p19q and IDH12 with intact ATRX.
IDH1/2 is mutated in most of diffuse astrocytoma, regardless of the grade, distinguishes astrocytoma with a more favorable course from IDH-wild type tumors which have a less favorable course [10].
In diffuse glioma cases of adults with unavailability of immunohistochemical ATRX and IDH1/2 antibodies as in our situation, 1p19q is first tested. If the tumor harbors 1p19q codeletion, no need to test for IDH1/2 as it should be mutated in adult oligodendroglioma. Only the cases with normal 1p19q, i.e., astrocytoma diagnosis molecular study for IDH1/2 should be requested.
Conclusions
OD carries a high risk of skeletal and extra- skeletal malignancy specially brain tumor. Our case we present OD with IDH-mutant diffuse astrocytoma. The previous literatures show that patients with OD develop gliomas at a relatively younger age and there is a high rate of multifocality [4, 5]. In addition, common locations are frontal lobe and brainstem.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
The authors declare that they have no conflict of interest.
Informed Consent
The patient has signed the informed consent.
Author Contributions
Assem Al Rumeh and Toleen Shaikh wrote the whole manuscript and provided the clinical information. Ahmad Lari did the surgery and provided the notes. Abdullah Muharib provided the radiology images with explanation of each image. Wafaa Al Shakweer reviewed the manuscript.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Kyritsis AP, Bondy ML, Rao JS, Sioka C. Inherited predisposition to glioma. Neuro Oncol. 2010;12(1):104-113.
doi pubmed - Silve C, Juppner H. Ollier disease. Orphanet J Rare Dis. 2006;1:37.
doi pubmed - Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F. World Health Organization classification of tumors. Pathology and genetics. Tumors of Soft Tissue and Bone. 4th Edition. 2013. Volume 5.
- Bathla G, Gupta S, Ong CK. Multifocal intracranial astrocytoma in a pediatric patient with Ollier disease. Indian J Radiol Imaging. 2012;22(1):58-62.
doi pubmed - Pearce P, Robertson T, Ortiz-Gomez JD, Rajah T, Tollesson G. Multifocal supratentorial diffuse glioma in a young patient with Ollier disease. J Clin Neurosci. 2012;19(3):477-478.
doi pubmed - Harrison RA, Fuller GN, Prabhu SS, Slopis JM, Penas-Prado M. Multicentric glioma in ollier disease: a case report and review of the literature. Neuro-Oncology. 2016;18(suppl_6):vi172.
doi - Khan SH, Rather TA, Koul PA, Makhdoomi R, Bhat AR, Malik D, Manohar R. Bone scintigraphy in Ollier's disease: A rare case report. Indian J Nucl Med. 2013;28(4):226-229.
doi pubmed - Hori K, Matsumine A, Niimi R, Maeda M, Uchida K, Nakamura T, Sudo A. Diffuse gliomas in an adolescent with multiple enchondromatosis (Ollier's disease). Oncol Lett. 2010;1(4):595-597.
doi pubmed - Gajavelli S, Nakhla J, Nasser R, Yassari R, Weidenheim KM, Graber J. Ollier disease with anaplastic astrocytoma: A review of the literature and a unique case. Surg Neurol Int. 2016;7(Suppl 23):S607-611.
doi pubmed - WHO classification of tumors of the central nervous system. 2016.
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Neurology Research is published by Elmer Press Inc.