

| Journal of Neurology Research, ISSN 1923-2845 print, 1923-2853 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Neurol Res and Elmer Press Inc |
| Journal website https://www.neurores.org |
Case Report
Volume 12, Number 1, February 2022, pages 21-24

A Diagnostic Mimicker: A Sixty-Year-Old Male With Ptosis and Left Arm Weakness
Connie Tanga, b, Sara Dehbashia
aDepartment of Neurology, Thomas Jefferson University Hospital, Philadelphia, PA, USA
bCorresponding Author: Connie Tang, Department of Neurology, Thomas Jefferson University Hospital, Philadelphia, PA, USA
Manuscript submitted June 12, 2020, accepted June 23, 2020, published online December 15, 2021
Short title: A Case With Ptosis and Left Arm Weakness
doi: https://doi.org/10.14740/jnr605
| Abstract | ▴Top |
We describe a case of a 60-year-old male presenting with ptosis and left arm weakness. We review key localization features, diagnostic studies, including electromyography (EMG) findings, and discuss treatment and prognosis of the pharyngeal-cervical-brachial (PCB) variant of Guillain-Barre syndrome. We explore the differential diagnoses from motor neuron disease, neuromuscular junction disorder, and other atypical Guillain-Barre syndromes that this condition can mimic and how to recognize the PCB variant in the neurologic evaluation.
Keywords: Neuromuscular disease; Clinical neurophysiology; EMG; Clinical neurology
| Introduction | ▴Top |
Guillain-Barre syndrome (GBS) is a heterogenous syndrome characterized by rapidly progressive polyneuropathy with weakness and areflexia. Several variations in this disease spectrum predominantly feature cranial neuropathies, such as Miller Fisher syndrome (MFS) that is a triad of ophthalmoplegia, gait ataxia, and areflexia, pharyngeal-cervical-brachial (PCB) variant presenting with bulbar dysfunction along with arm and neck weakness, and intersection of these two variant syndromes known as Fisher-pharyngeal-cervical-brachial overlap syndrome. We present a case that reflects key neurologic differential diagnoses that may mimic this condition and the importance of prompt recognition to provide appropriate treatment, which may support the role of serial intravenous immunoglobulin (IVIG) treatment.
| Case Report | ▴Top |
Investigations
A 60-year-old man with a past medical history of hypertension and hyperlipidemia acutely presented with left facial droop with concurrent dysarthria and dysphagia along with left arm weakness. A day later, he developed bilateral ptosis, left more predominant than right. Magnetic resonance imaging (MRI) brain with and without contrast and magnetic resonance angiography (MRA) head obtained at a local hospital were unremarkable.
A week prior to admission, he developed nasal congestion and a cough that was improving. He denied any headache, visual changes, anosmia or dysgeusia. On presentation, vital signs were stable. Neurologic exam revealed equally reactive pupils, left facial weakness with no forehead wrinkling, weak cough, lingual dysarthria, neck flexion weakness (4+/5 on MRC scale), bilateral shoulder elevation weakness (4/5), and left shoulder abduction weakness (4/5) along with areflexia throughout all extremities.
Questions for consideration at that time included: 1) Would this lesion localize to the central nervous system (CNS) or peripheral nervous system (PNS)? 2) What would be in the differential diagnosis? 3) What tests would help narrow the differential diagnosis?
Diagnosis
Localizing a lesion that affects multiple cranial nerves can encompass the brainstem, neuromuscular junction (NMJ), or be a separate peripheral nerve process. In this case, the patient has cranial neuropathies of nerves 3, 7, 9, 10, 11 and 12. As there is paralysis of the frontalis muscle, the facial weakness is consistent with a peripheral cranial nerve 7 lesion, which can originate from the facial nucleus in the pons and later divides into five peripheral branches innervating the muscles of facial expression. This is further supported by an MRI brain study without brainstem lesions, as may be seen in stroke or rhombencephalitis. Additionally, involvement of the neck flexors, trapezius, and deltoid muscles is more in favor of a peripheral nervous system disorder.
NMJ disorders include consideration of myasthenia gravis (MG), Lambert-Eaton myasthenic syndrome (LEMS), and botulism. MG presents in a bimodal distribution of young women and older men, and symptoms commonly present with bilateral ptosis, bulbar dysfunction, and neck flexion and proximal shoulder weakness. LEMS incidence is rarer than MG, but should be considered in an older adult when symptoms present insidiously with proximal weakness although involvement of the oropharyngeal and ocular muscles is to a lesser degree. Botulism is a rare but life-threatening neuroparalytic syndrome that presents with cranial neuropathies and descending weakness. The patient’s lack of environmental and food exposure, gastrointestinal symptoms, and normal pupillary response is less supportive.
Evaluation for MG includes serum studies for acetylcholine receptor (AChR) antibodies and muscle specific-kinase (MuSK) antibodies, which were negative for this patient. Effectively, this suggests a lower likelihood that this presentation is MG. Although there are seronegative forms of the disorder, we should consider other possibilities such as GBS.
GBS is a heterogeneous syndrome characterized by rapidly progressive polyneuropathy with weakness and areflexia. Several variations in this disease spectrum predominantly feature cranial neuropathies, such as MFS, PCB variant, and intersection of these two variant syndromes known as Fisher-pharyngeal-cervical-brachial overlap syndrome. Diagnosis is supported by albuminocytologic dissociation seen in cerebrospinal fluid (CSF) analysis although protein elevation may not be appreciated in the first week by one-third of patients. Our patient had four white blood cells (WBCs)/µL with mildly elevated protein at 49 mg/dL (normal 45 mg/dL). Additional serum markers for ganglioside antibodies are supportive, such as anti-GQ1b associated with MFS, and anti-GT1a and anti-GD1b both associated with PCB variant. Ganglioside antibodies assessed in our patient were negative.
Treatment
At this juncture, electrodiagnostic studies can assist in localizing the lesion to the NMJ or peripheral nerves and characterize the degree of nerve injury. Due to lack of accessibility for an inpatient study and worsening dysphagia leading to placement of a nasogastric tube for nutrition, empiric immunomodulatory therapy was initiated. As IVIG is a first-line agent in both myasthenic crisis and GBS, a dose of 2 g/kg was given over the course of 3 days. The patient’s dysphagia improved to avoid percutaneous endoscopic gastrostomy, and he was discharged to inpatient rehabilitation.
Follow-up and outcomes
One month after hospitalization, his speech improved but retained a nasal quality, along with resolution of left ptosis and facial droop. He graduated from pureed foods to soft-textured foods over 2 weeks. He reported saliva pooling, but not significant enough to cause excessive drooling or choking. Initial nerve conduction study (NCS) revealed conduction block of the proximal median nerve (Fig. 1) as well as slightly prolonged distal motor latencies in the peroneal and tibial nerves with preserved compound muscle action potential (CMAP) amplitudes and normal conduction velocities. Respective peroneal and tibial F-responses were absent. Sensory conduction studies were normal. EMG study revealed 1+ to 3+ spontaneous activity in the cranial and cervical segments with neurogenic appearing motor unit action potentials whereas thoracic and lumbosacral segments did not demonstrate any denervation.
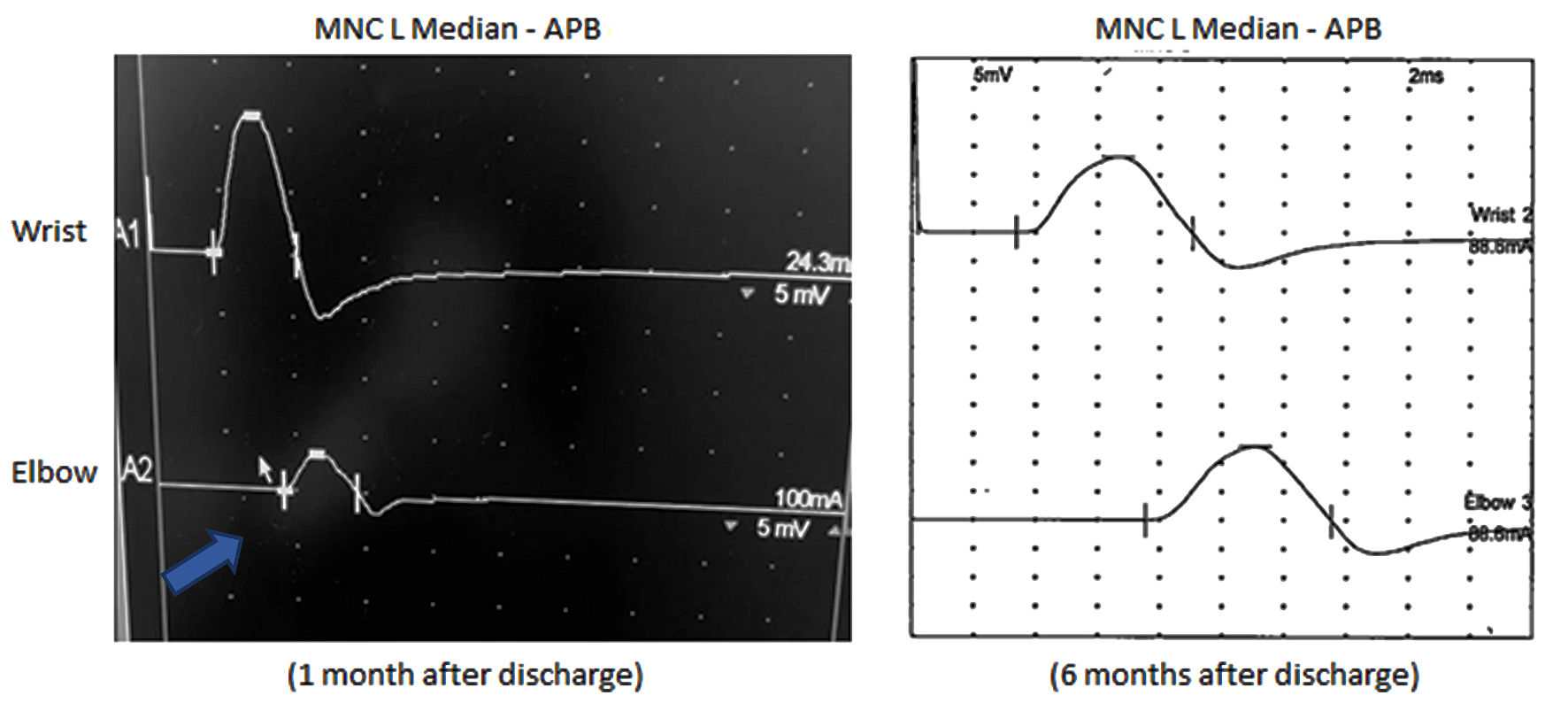 Click for large image | Figure 1. Left median motor nerve conduction (MNC) study. Conduction block (arrow) is demonstrated on the left screen with more than 50% decrease in CMAP amplitude at proximal site (elbow 3.3 mV) when compared to distal site (wrist 11.1 mV). The right screen shows resolution of conduction block after several months of IVIG (CMAP amplitude at wrist (13.2 mV) and elbow (12.8 mV)). There is also improvement of motor distal latency onset from 4.6 ms to 3.4 ms over the 6-month period. CMAP: compound muscle action potential; IVIG: intravenous immunoglobulin; APB: abductor pollicis brevis. |
Questions for consideration were: 1) What other diagnosis is concerning with diffuse active denervation? 2) What additional studies should be performed? 3) What further treatment options are available?
While acute bulbar palsy can be a presenting symptom in stroke and MG, progressive bulbar palsy, a motor neuron disease subtype, begins as a several-month history of worsening dysarthria with gagging, choking, and weight loss. Speech is slow and spastic with variable flaccid features. The tongue may be atrophied with fasciculations accompanied by brisk facial reflexes. While a majority of cases evolve to ALS, occasionally symptoms remain relatively restricted to the bulbar muscles. In this patient’s case, his electrodiagnostic study showed mixed axonal and demyelinating features, and without sensory nerve involvement. Arguably, finding of conduction block of the proximal median nerve and absence of wide spread active denervation involving lower and upper extremities would signify that the underlying disorder is a motor neuropathy and not a motor neuron disease.
As the patient was ambulatory but with persistent speech impairment, consideration was given for an additional three series of IVIG, in which the patient demonstrated continued improvement of his tongue weakness and speech. He returned to eating food without modified consistency, and his left shoulder strength significantly improved to where he was able to participate in skiing. He also underwent 3 months of speech therapy and persisted with oral exercises afterwards, which must also be given credit towards his betterment. Repeat EMG study 6 months after the initial study shows resolution of active denervation, conduction block, and prolonged distal latencies.
| Discussion | ▴Top |
With improved clinical symptoms and resolving electrodiagnostic findings, especially after treatment with IVIG, we conclude that the patient’s diagnosis is most fitting with an acute to subacute presentation of an acquired inflammatory axonal polyradiculoneuropathy of the PCB variant.
PCB variant is a localized subtype of GBS with rapidly progressive oropharyngeal and cervicobrachial weakness associated with areflexia in the upper limbs with strength in the lower extremities usually preserved [1]. There are presentations that may not demonstrate all features as there is even an acute bulbar palsy without limb weakness that is described as a continuum with either MFS or PCB [2]. A prospective study of 250 patients revealed that PCB (3%) was the second most frequent GBS variant [3]. The largest case series examined 100 patients with PCB revealing median age at 43 years, a slight male predominance, and antecedent upper respiratory tract infection (70%) or diarrhea (30%) [4]. Approximately two-thirds of patients in the series of Nagashima et al were also noted to have paresthesias in the upper extremities with few (10%) having normal or exaggerated reflexes [5].
The suggested diagnostic criteria have evolved to include sensory disturbances and preserved reflexes, and highlight an acute and monophasic disorder with supportive evidence of an antecedent illness, CSF albuminocytological dissociation, electrodiagnostic correlation of neuropathy, and/or detection of immunoglobulin G (IgG) anti-GT1a or anti-GQ1b antibodies [4]. Presence of ataxia or altered consciousness may prompt an alternative diagnosis, such as PCB overlapping with MFS or Bickerstaff encephalopathy.
As with other GBS variants, autoantibodies against neuronal gangliosides have been identified in the pathogenesis of the disease, and it is postulated that the nature of the epitope as a glycolipid, shows molecular similarity between the myelin epitopes and glycolipids expressed on Campylobacter, Mycoplasma, cytomegalovirus (CMV), and other infectious agents [6]. Half of patients with PCB carry IgG GT1a antibodies, which are more strongly expressed in the glossopharyngeal and vagal nerves than GQ1b antibodies, whereas both antibodies were similarly expressed in the oculomotor nerves [1]. Although our patient did not test positive for a ganglioside antibody, there are other axonal serological markers that may be outside of the scope of the panel tested with other reports of IgG antibodies to GM1, GM1b, GD1a, or GalNAc-GD1a seen in PCB [5]. During the coronavirus disease 2019 (COVID-19) pandemic, there have been several case reports of associated GBS, and GD1b antibody found in association with MFS [7].
PCB can also be representative of a focal form of acute motor axonal neuropathy (AMAN) based on electrodiagnostic testing. In Ropper’s original case series, NCS did not reveal apparent demyelinating features such as temporal dispersion with follow-up testing showing rapid recovery with no implication of remyelination [8]. Conduction blocks can be determined in 67% of AMAN patients, and in one retrospective study, unobtainable F-responses were seen in three patients with AMAN [9, 10].
Therapy is well established in GBS with the use of IVIG or plasmapheresis with the latter often employed in those with severe paralysis and/or respiratory failure. Based on two class I studies, results demonstrated that IVIG is at least as effective as plasmapheresis [11, 12]. Coupled with ease of access and shorter duration, this perhaps accounts for its more prevalent use as a first-line agent. However, sequential treatment using plasma exchange followed by IVIG offers no additional benefit. Due to bulbar involvement, prompt recognition and early treatment can help avoid worsening dysphagia and respiratory involvement that may lead to nasogastric feeding and mechanical ventilation. Although GBS and its variants are considered monophasic illnesses, there is an inclination for repeat courses of IVIG in those with poorer prognosis. As there has been observational evidence to both support and refute effectiveness of a second course of IVIG in reducing morbidity, it remains a clinical decision to be determined on an individual basis until there are further prospective randomized trials [13, 14]. Our patient continued to derive benefit with additional IVIG therapies and is monitored every 3 months to assess for dependency.
Learning points
The clinical phenotype of PCB is often underrecognized. However, with prompt identification and knowledge of critical differential diagnoses, there may be more accurate characterization of this subtype that will allow for earlier treatment and better prognosis as well as consideration for repeated IVIG therapy in patients with protracted recovery.
Acknowledgments
None to declare.
Financial Disclosure
The authors report no relevant disclosures or funding.
Conflict of Interest
The authors report no conflict of interest.
Informed Consent
The patient in the case has provided written consent for publication.
Author Contributions
Connie Tang and Sara Dehbashi equally contributed to writing the manuscript.
Data Availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
| References | ▴Top |
- Koga M, Yuki N, Ariga T, Morimatsu M, Hirata K. Is IgG anti-GT1a antibody associated with pharyngeal-cervical-brachial weakness or oropharyngeal palsy in Guillain-Barre syndrome? J Neuroimmunol. 1998;86(1):74-79.
doi - Kim JK, Kim BJ, Shin HY, Shin KJ, Nam TS, Oh J, Suh BC, et al. Acute bulbar palsy as a variant of Guillain-Barre syndrome. Neurology. 2016;86(8):742-747.
doi pubmed - Ropper AH. Severe acute Guillain-Barre syndrome. Neurology. 1986;36(3):429-432.
doi pubmed - Wakerley BR, Yuki N. Pharyngeal-cervical-brachial variant of Guillain-Barre syndrome. J Neurol Neurosurg Psychiatry. 2014;85(3):339-344.
doi pubmed - Nagashima T, Koga M, Odaka M, Hirata K, Yuki N. Continuous spectrum of pharyngeal-cervical-brachial variant of Guillain-Barre syndrome. Arch Neurol. 2007;64(10):1519-1523.
doi pubmed - Amato AA, Russell JA. Guillain-Barre syndrome and related disorders. In: Neuromuscular Disorders, 2nd ed. McGraw Hill. 2008; p. 320-325.
- Gutierrez-Ortiz C, Mendez-Guerrero A, Rodrigo-Rey S, San Pedro-Murillo E, Bermejo-Guerrero L, Gordo-Manas R, de Aragon-Gomez F, et al. Miller Fisher syndrome and polyneuritis cranialis in COVID-19. Neurology. 2020;95(5):e601-e605.
doi pubmed - Ropper AH. Further regional variants of acute immune polyneuropathy. Bifacial weakness or sixth nerve paresis with paresthesias, lumbar polyradiculopathy, and ataxia with pharyngeal-cervical-brachial weakness. Arch Neurol. 1994;51(7):671-675.
doi pubmed - Uncini A, Manzoli C, Notturno F, Capasso M. Pitfalls in electrodiagnosis of Guillain-Barre syndrome subtypes. J Neurol Neurosurg Psychiatry. 2010;81(10):1157-1163.
doi pubmed - Yadegari S, Nafissi S, Kazemi N. Comparison of electrophysiological findings in axonal and demyelinating Guillain-Barre syndrome. Iran J Neurol. 2014;13(3):138-143.
- van der Meche FG, Schmitz PI. A randomized trial comparing intravenous immune globulin and plasma exchange in Guillain-Barre syndrome. Dutch Guillain-Barre Study Group. N Engl J Med. 1992;326(17):1123-1129.
doi pubmed - The North American study of plasmapheresis in the Guillain-Barre syndrome. The Guillain-Barre' Syndrome Study Group. J Clin Apher. 1985;2(4):315-320.
doi pubmed - Verboon C, van den Berg B, Cornblath DR, Venema E, Gorson KC, Lunn MP, Lingsma H, et al. Original research: Second IVIg course in Guillain-Barre syndrome with poor prognosis: the non-randomised ISID study. J Neurol Neurosurg Psychiatry. 2020;91(2):113-121.
doi pubmed - Godoy DA, Rabinstein A. Is a second cycle of immunoglobulin justified in axonal forms of Guillain-Barre syndrome? Arq Neuropsiquiatr. 2015;73(10):848-851.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Neurology Research is published by Elmer Press Inc.