

| Journal of Neurology Research, ISSN 1923-2845 print, 1923-2853 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Neurol Res and Elmer Press Inc |
| Journal website https://www.neurores.org |
Case Report
Volume 11, Number 1-2, April 2021, pages 32-36
Thymic Neoplasm: A Rare Disease With Unusual Neurologic Manifestations
Sara Esmaeilia, b, c, Seyedeh Niloufar Rafiee Alavib, c, Sevim Soleimanid, Mohammad Mojtahedb, Mahshid Panahie, Yalda Nilipourf, Bahram Haghi Ashtianib, g
aCellular and Molecular Research Centre, Iran University of Medical Sciences, Tehran, Iran
bDepartment of Neurology, Iran University of Medical Sciences, Tehran, Iran
cStudent Research Committee, Iran University of Medical Sciences, Tehran, Iran
dSchool of Medicine, Shahid Beheshti Medical University, Tehran, Iran
ePathology Department, Iran University of Medical Sciences, Tehran, Iran
fPediatric Pathology Research Center, Research Institute for Children’s Health, Shahid Beheshti University of Medical Sciences, Tehran, Iran
gCorresponding Author: Bahram Haghi Ashtiani, Department of Neurology, Iran University of Medical Sciences, Tehran, Iran
Manuscript submitted August 14, 2020, accepted October 17, 2020, published online April 9, 2021
Short title: A Rare Disease of Thymic Neoplasm
doi: https://doi.org/10.14740/jnr625
| Abstract | ▴Top |
Thymoma is a rare tumor that is commonly associated with autoimmune diseases. Of these, myasthenia gravis (MG) is widely considered as the most common paraneoplastic condition. On the other hand, dermatomyositis (DM) has been rarely reported as the first presentation of non-invasive thymoma. Hereby, we describe a patient with non-invasive thymoma who initially presented with painful symmetric proximal muscle weakness with no signs of ptosis or diplopia. The needle electromyography (EMG) revealed spontaneous/insertional activity in proximal muscles. The patient was finally diagnosed with DM by muscle biopsy. Spiral chest computed tomography (CT) scan coupled with pathological assessment confirmed a non-invasive thymoma with diffuse reaction for P63 and pancytokeratin (Panck). Corticosteroids pulse therapy was initiated and the patient was referred for thymectomy. A few months later, patient began to display other neurological symptoms such as ptosis and diplopia with fluctuating pattern. As coexistence of MG was presumed, nerve conduction study (NCS) study was performed and slow repetitive nerve stimulation in proximal muscles showed more than 10% decrement in compound muscle action potential (CMAP) amplitude in repetitive nerve stimulation (RNS). Further workup revealed a positive anti-acetylcholine receptor antibody with high titer. Thus, MG was confirmed. No more treatment options were planned. Low dose corticosteroids were continued and azathioprine and pyridostigmine were prescribed. During follow-ups, symptoms were fully controlled.
Keywords: Thymoma; Myasthenia gravis; Dermatomyositis
| Introduction | ▴Top |
Thymoma is a rare tumor that is commonly associated with autoimmune diseases as it induces autoantibody formation, particularly against neuromuscular system. Myasthenia gravis (MG) is well known as the first presentation of thymoma [1] Dermatomyositis (DM), on the other hand is rarely reported as the first presentation of non-invasive thymoma [1-3]. Also, initial presentations of MG after thymectomy has been rarely reported [1]. Finally, DM and MG are both autoimmune disorders presenting with muscle weakness, which scarcely occur simultaneously in the same patient and the diagnosis and differentiation might not be straightforward [4-6]. Here we describe a patient with non-invasive thymoma with primary presentation of DM who subsequently developed signs of MG after thymectomy. It is important for neurologists to be conscious of presence of both diseases as the paraneoplastic syndromes in patients who have thymoma, since the treatment of MG may differ from that of myositis. Also, it is worth mentioning that this coexistence provides important information about complex pathophysiology of thymoma associated immune disorders.
| Case Report | ▴Top |
A 35-year-old female was admitted with symmetric proximal muscle weakness and pain which had started 3 weeks prior to the symptoms. She described the symptoms to gradually involve the upper extremities with no fluctuations. She had complaints of malaise, fatigue and weight loss of 9 kg in 4 months accompanied with loss of appetite. Sensory complaints were absent. Bulbar symptoms including dysphagia were limited to solid foods and dysphonia was noticed. However, she had no history of ptosis or diplopia. Family history of the patient was not significant for periodic paralysis, muscular dystrophies; and she had no history of specific corresponding medications or occupational exposures. However, the patient had a history of autoimmune hypothyroidism (positive anti-thyroid peroxidase (TPO)).
On physical examination, ptosis was not apparent and skin change was unremarkable. Except for neck flexor muscle weakness, Cranial nerve examination was intact. Motor examinations revealed quadriparesis involving proximal musculature as positive Gowers’ sign. However, atrophy was not prominent and distal strength was normal. Sensory examination, tendon reflexes, cerebellar examination and gait examination were all unremarkable. The patient had elevated creatine kinase (CPK = 5,560), aspartate aminotransferase and alanine aminotransferase (ALT = 435, AST = 330). Anti-TPO was positive. Nerve conduction study (NCS) findings were unremarkable. The needle electromyography (EMG) revealed spontaneous/insertional activity in proximal muscles. At the time, the motor unit action potential (MUAP) waveform became small, polyphasic with sort duration, and abnormal interference patterns were found in proximal muscles. As a result, EMG was compatible with myopathy of proximal muscles with denervation features, suggesting inflammatory polymyopathy (Table 1). Muscle biopsy showed severe perifascicular atrophy and necrosis with extensive myophagocytosis and chronic inflammation (Fig.1). On further workups, enzyme-linked immunoassay (ELISA)-based anti-Jo1, anti-serine protease 3 (PR3) and anti-myeloperoxidase (MPO) immunoassays (EIAs) were all negative. Rheumatologic markers were all normal except for antinuclear antibody (ANA), which was weakly (1/40) positive with speckled pattern. The diagnosis of DM was confirmed by current international guidelines [7, 8].
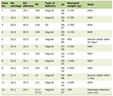 Click to view | Table 1. EMG Summary |
 Click for large image | Figure 1. Muscle biopsy. Severe perifascicular atrophy and necrosis with extensive myophagocytosis and chronic inflammation (hematoxylin and eosin (H&E), × 100). |
Additionally, workup for underlying malignancy revealed all tumor markers to be negative, unless a spiral chest computed tomography (CT) scan, which illustrated a solid, lobulated, well defined and homogenous mass in anterior mediastinum with size of 60 × 43 × 90 mm (Fig. 2). The pathology confirmed a non-invasive thymoma with diffuse reaction for P63 and pancytokeratin (Panck) (Fig. 3). Corticosteroids pulse therapy was initiated and the patient was referred for thymectomy. During treatment with low-dose corticosteroids as maintenance therapy, the patient began to display other neurological symptoms such as ptosis and diplopia with fluctuating pattern. A few months later, based on increased bulbar complaints and fluctuating fatigable weakness, coexistence of MG was presumed. Subsequent NCS study was performed and slow repetitive nerve stimulation in proximal muscles, including trapezius showing more than 10% decrement in compound muscle action potential (CMAP) amplitude in repetitive nerve stimulation (RNS) (Fig. 4). Workups revealed a positive anti-acetylcholine receptor antibody with high titer. Considering abovementioned findings, MG was confirmed based on current international guidelines [9].
 Click for large image | Figure 2. Spiral chest computed tomography (CT) scan. The arrow on spiral chest CT scan illustrates a solid, lobulated, well defined and homogenous mass in anterior mediastinum with size of 60 × 43 × 90 mm. |
 Click for large image | Figure 3. Thymoma. (a) Cellular lobulated architecture lay on thick fibrous band (hematoxylin and eosin (H&E), × 40). (b) Predominant lymphocytes and scattered polygonal epithelial cells (H&E, × 400). (c) Pancytokeratin (Panck) highlights diffuse mesh work of tumoral cells (× 400). |
 Click for large image | Figure 4. Repetitive nerve stimulation (RNS). Slow RNS in trapezius demonstrates more than 10% decrement in compound muscle action potential (CMAP) amplitude. |
After consulting with an oncologist, it was chosen not to pursue any further cancer treatment options, including chemotherapy or radiotherapy. However, slow-dose corticosteroids along with azathioprine and pyridostigmine were prescribed. During follow-ups, the patient felt much better and symptoms were fully controlled.
| Discussion | ▴Top |
This is a report of a patient whose primary presentation of non-invasive thymoma was indicative of DM but subsequently developed to MG.
Thymoma is associated with paraneoplastic syndromes (PNDs). Diagnosis of these disorders is not straightforward and usually needs specific autoantibodies. Moreover, coexistence of MG and myositis as paraneoplastic disorders, especially DM being prior, is very rare [1]. As a result, it could be challenging for the clinicians to suspect other underlying causes. To date, there have been limited reports of DM-associated thymoma [3], and even less reports of patients who developed both MG and myositis simultaneously [1]. It is also noticeable that in these reports, similar to our case, the complaints of diplopia or ptosis along with muscle weakness were present before thymectomy, and not after [4-6].
An interesting point in this case is that diplopia and ptosis are classic features of MG and are not usually seen in patients with myositis alone [4, 10]. Yet, our patient had never experienced ptosis or diplopia before thymectomy, and the symptoms occurred during corticosteroids therapy. Nevertheless, corticosteroid induced myopathy could be a possible explanation. However, this diagnosis was ruled out by second electrodiagnostic (EDX) along with the high titer of acetylcholine receptor (AChR) antibody.
The fatigability and increased bulbar symptoms were the only clues which guided us towards diagnosing MG. Fatigability is not characteristic of myositis but it is a very common symptom of the other disorder [10]. Meanwhile, fatigability with fluctuating weakness and bulbar symptoms such as dysphagia and dysarthria are characteristic of both myositis and MG [4]. Our final diagnosis for each disease was made both by EMG and biopsy results. The pivotal point is that although the prevalence of bulbar symptoms is less than 40% in myositis, about two-thirds of MG patients have bulbar symptoms [4, 11].
Non-invasive thymoma was found as the underlying pathology for concurrence of DM and MG. As abovementioned, thymoma-associated autoimmune diseases have been introduced [12].These autoimmune disorders could be categorized as thymoma-associated paraneoplastic diseases [2]. These include MG as the most common diagnosis encephalitis, acquired neuromyotonia (Isaacs’ syndrome), Morvan’s syndrome myositis and finally, anti-PIT-1 antibody syndrome [2, 12]. These disorders can cause severe disabilities and management of these disorders should include a comprehensive malignancy evaluation for possible underlying thymoma. Efficient management should include tumor resection along with appropriate management of the autoimmune conditions such as using corticosteroids. It has been found that these paraneoplastic disorders mainly respond well to treatment of thymoma [2, 12].
Thymus is the key organ for regulating immune tolerance. T cells migrate and mature in thymic cortex and medulla. This happens via somatic gene rearrangement of T cells. These T cells express diverse range of receptors during development. During this process the cells with excess reactivity to self-antigens are not normally allowed to survive. Dysregulation in this process could result in later autoimmune complications [2].
In thymoma-associated MG, the patients’ thymus has maintained its capability of releasing mature T cells. But neoplastic epithelial cells express epitopes of AChR subunits and titin. Also, they express lower levels of major histocompatibility class II molecules and autoimmune regulator gene compared to normal cells. This leads to imperfect negative selection, which results in autoreactive CD4+ T cells to be released, appearing to be the main cause of thymoma-associated MG. However, the exact pathophysiology is yet unknown. Accordingly, the cause of such coexistence of DM and MG with underlying thymoma is still in debate.
Future investigations, such as discovering more novel autoantibodies, can accelerate the final diagnosis, especially in “seronegative” cases. Additionally, it appears that the role of some of these antibodies is still unclear and much is yet to be learned to fully understand the mechanism of this disease and its potential treatments [2, 13].
Conclusions
DM may be the primary presentation of thymoma and MG might become symptomatic later in time and even after thymectomy. We suggest that any prominent bulbar symptoms along with fatigable weakness, even without ptosis and diplopia, must raise the suspicion of possibility of concurrent MG in patients with myositis and should be confirmed through appropriate electrophysiologic studies and autoantibody tests.
Acknowledgments
The authors would like to thank Ms. Maryam Daneshgar for the English language editing and Dr. Peyman Shirani for his kind support.
Financial Disclosure
Non to declare.
Conflict of Interest
None to declare.
Informed Consent
Written consent was obtained from the patient.
Author Contributions
SE, SNRA, SS, MM, MP, YN and BHA designed and performed the clinical and/or paraclinical experiments, and co-wrote the paper. MP, YN and BHA supervised the research.
Data Availability
Any inquiries regarding supporting data availability of this study should be directed to the corresponding author.
| References | ▴Top |
- Klein JJ, Gottlieb AJ, Mones RJ, Appel SH, Osserman KE. Thymoma and polymyositis. onset of myasthenia gravis after thymectomy: report of two cases. Arch Intern Med. 1964;113:142-152.
doi pubmed - Evoli A, Lancaster E. Paraneoplastic disorders in thymoma patients. J Thorac Oncol. 2014;9(9 Suppl 2):S143-147.
doi pubmed - Ago T, Nakamura M, Iwata I, Murai H, Okuma K, Tsuru T, Kaji Y, et al. Dermatomyositis associated with invasive thymoma. Intern Med. 1999;38(2):155-159.
doi pubmed - Paik JJ, Corse AM, Mammen AL. The co-existence of myasthenia gravis in patients with myositis: a case series. Semin Arthritis Rheum. 2014;43(6):792-796.
doi pubmed - Sanguesa Gomez C, Flores Robles BJ, Mendez Perles C, Barbadillo C, Godoy H, Andreu JL. Dermatomyositis and myastenia gravis: An uncommon association with therapeutic implications. Reumatol Clin. 2015;11(4):244-246.
doi - Santos E, Coutinho E, Martins da Silva A, Marinho A, Vasconcelos C, Taipa R, Pires MM, et al. Inflammatory myopathy associated with myasthenia gravis with and without thymic pathology: Report of four cases and literature review. Autoimmun Rev. 2017;16(6):644-649.
doi pubmed - Miller FW. New approaches to the assessment and treatment of the idiopathic inflammatory myopathies. Ann Rheum Dis. 2012;71(Suppl 2):i82-85.
doi pubmed - Raychaudhuri SP, Mitra A. Polymyositis and dermatomyositis: Disease spectrum and classification. Indian J Dermatol. 2012;57(5):366-370.
doi pubmed - Hehir MK, Silvestri NJ. Generalized myasthenia gravis: classification, clinical presentation, natural history, and epidemiology. Neurol Clin. 2018;36(2):253-260.
doi pubmed - Strowd LC, Jorizzo JL. Review of dermatomyositis: establishing the diagnosis and treatment algorithm. J Dermatolog Treat. 2013;24(6):418-421.
doi pubmed - Benatar M. A systematic review of diagnostic studies in myasthenia gravis. Neuromuscul Disord. 2006;16(7):459-467.
doi pubmed - Bando H, Iguchi G, Okimura Y, Odake Y, Yoshida K, Matsumoto R, Suda K, et al. A novel thymoma-associated autoimmune disease: Anti-PIT-1 antibody syndrome. Sci Rep. 2017;7:43060.
doi pubmed - Mantegazza R, Bernasconi P, Cavalcante P. Myasthenia gravis: from autoantibodies to therapy. Curr Opin Neurol. 2018;31(5):517-525.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Neurology Research is published by Elmer Press Inc.