

| Journal of Neurology Research, ISSN 1923-2845 print, 1923-2853 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Neurol Res and Elmer Press Inc |
| Journal website http://www.neurores.org |
Case Report
Volume 7, Number 4-5, October 2017, pages 85-88
Chordoid Glioma of Third Ventricle Presenting as Primary Polydipsia With Aggressive Behavior: A Rare Case With Rare Presentation
Abhinav Kumar Guptaa, d, Deepak Chand Guptaa, Saqib Ahmad Khana, Mohd. Razi Syedb, Puneet Guptac, Keshav Kumar Guptaa
aDepartment of Endocrinology, L.L.R.M. Medical College, Meerut, Uttar Pradesh, India
bDepartment of Endocrinology, Sri Sai Hospital, Muradabad, Uttar Pradesh, India
cDepartment of Medicine, Rukmani Birla Devi Hospital, Jaipur, Rajsthan, India
dCorresponding Author: Abhinav Kumar Gupta, Department of Endocrinology, L.L.R.M. Medical College, Meerut, India
Manuscript submitted April 4, 2017, accepted May 8, 2017
Short title: Chordoid Glioma of Third Ventricle
doi: https://doi.org/10.14740/jnr424e
| Abstract | ▴Top |
Chordoid glioma of third ventricle is relatively rare and characterized by unique histomorphology and varied clinical presentation. Chordoid glioma has high morbidity and high mortality rates despite being low grade tumor because of insidious presentation and difficulty in complete resection. Here, we report a new case of chordoid glioma occurring in a 32-year-old male, who presented with polyuria, polydipsia and behavioral problems, and on the water deprivation test diagnosis of primary polydipsia was made. MRI revealed tumor of third ventricle, which was resected subtotally. Diagnosis of chordoid glioma was confirmed by histology and immunohistochemical analysis of resected tumor. Recurrence of chordoid glioma was reported on MRI after 2.5 months. Based on this case report, we suggest chordoid glioma should be kept in differential diagnosis of primary polydipsia.
Keywords: Chordoid glioma; Primary polydipsia; Hypothalamic dysfunction; Endocrine dysfunction
| Introduction | ▴Top |
Chordoid glioma is a rare low grade tumor that arises from the anterior wall or roof of the third ventricle. Brat et al in 1998 described a series of eight cases of third ventricular neoplasm with a distinctive chordoid appearance that appears to represent a separate clinicopathological entity and named it chordoid glioma [1]. World Health Organization (WHO) classified it grade II neuroepithelial tumors of uncertain histogenesis [2]. It usually occurs in adults (median age at diagnosis, 48 years) with a female predominance of 2:1 [3].
This tumor was named chordoid glioma because of its distinct histologic appearance, reminiscent of chordoma, and its avid staining with glial fibrillary acid protein (GFAP) in immunohistochemical analysis [4].
Chordoid glioma usually presents with headache, visual disturbances and memory deficit, less frequent presentation is with endocrine disturbances (amenorrhea, hypothyroidism, weight gain, polyuria and polydipsia), and other focal deficits are rare [3].
We report a case of chordoid glioma presenting with primary polydipsia and aggressive behavior.
| Case Report | ▴Top |
We present a case of a 32-year-old male, who was admitted to our hospital for evaluation of polyuria (8 L/day), polydipsia and behavioral problem. He was not having any history of headache, visual disturbances and memory deficit. Visual acuity, visual field and other neurological examination were within normal limit.
Water deprivation test was performed after obtaining baseline values. His baseline sodium was 116.6 mEq/L, serum osmolality was 241 mOsm/kg of H2O and urine osmolality was 48 mOsm/kg of H2O. On water deprivation test urine osmolality increased to reach a value of 684 mOsm/kg of H2O. Serum calcium, potassium, glucose, cortisol, testosterone, gonadotropins and thyroid function test levels were within normal limit.
A magnetic resonance imaging (MRI) scan revealed a large lobulated, solid mass of 3.8 × 3.9 × 3.7 cm size in suprasellar and right parasellar region, isointense on T1W and hyperintense on T2W, abutting the optic chiasma and tracts (Fig. 1a-c). Provisional diagnosis of chordoid glioma of third ventricle or optico-chiasmatic hypothalamic glioma was made.
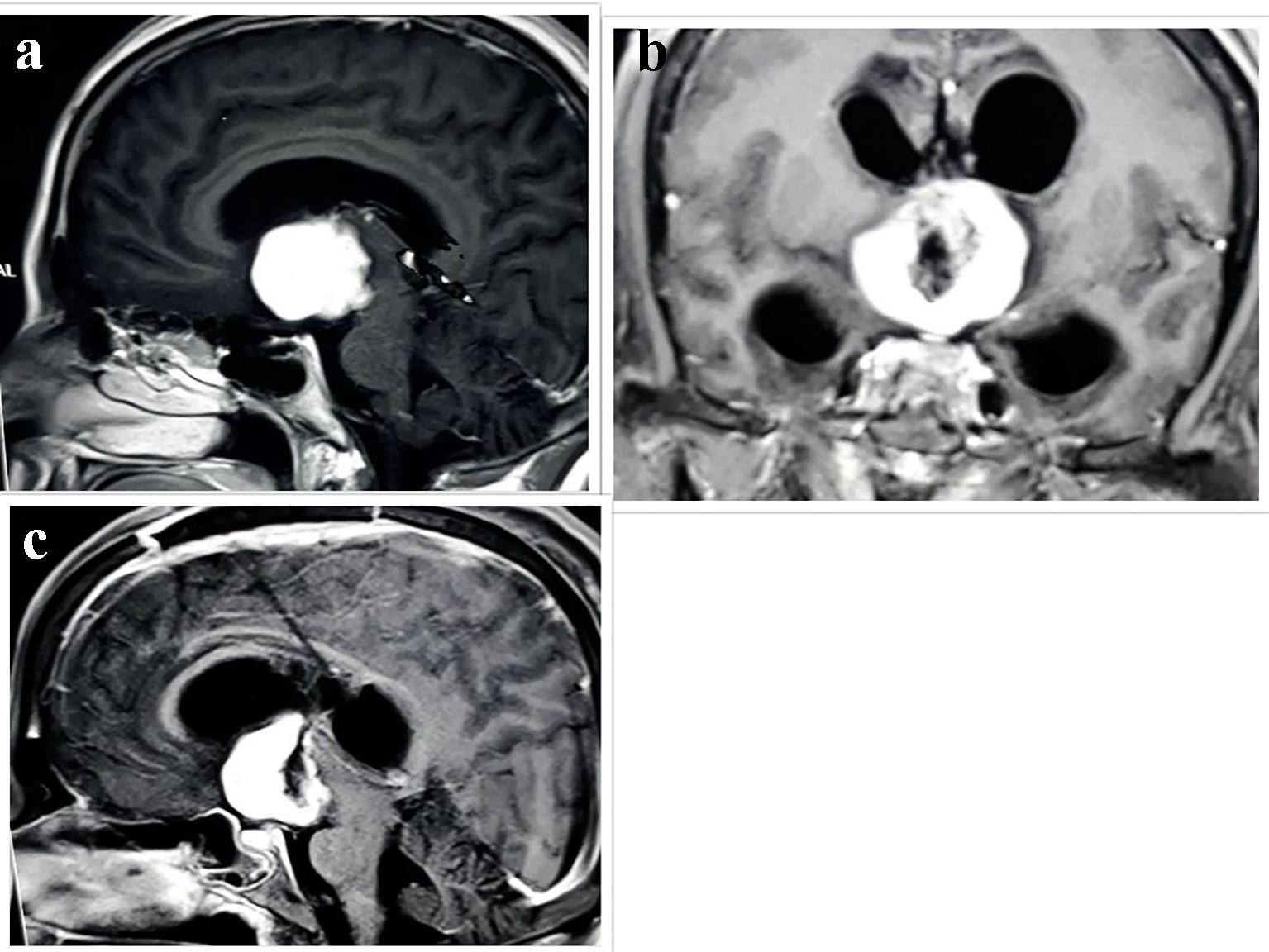 Click for large image | Figure 1. Radiological evaluation. (a) T1W MRI scan showing isointense lesion with homogenous enhancement in suprasellar and right parasellar region. Postoperative MRI scan after 2.5 months in (b) coronal and (c) sagittal scan showing heterogeneously enhancing hypointense mass lesion in suprasellar cistern and anterior part of third ventricle. |
Subsequently right frontal craniotomy via transcallosal, transventricular approach revealed the third ventricular mass adhering to surrounding structures. Hence the tumor was subtotally removed. Postoperative computed tomography (CT) showed remnant enhanced lesion.
Histopathological examination of tumor tissue revealed cluster and cords of epitheliod tumor cells in variable mucinous stroma. Tumor cells were round to oval uniform nucleus and abundant eosinophilic cytoplasm. Few bipolar cells and foci of lymphoplasmacytic infiltrate were seen. No mitoses, microvascular proliferation or necrosis were seen. Alcian blue stain was positive in mucinous stroma. On immunohistochemistry, tumor cells showed diffuse GFAP positivity and patchy CD34 positivity in aggregates of tumor cells. Epithelial membrane antibody (EMA) marked plasma cells and was negative in tumor cells (Fig. 2a-d). Ki-67 proliferation index was around 1%. Final diagnosis of chordoid glioma was made.
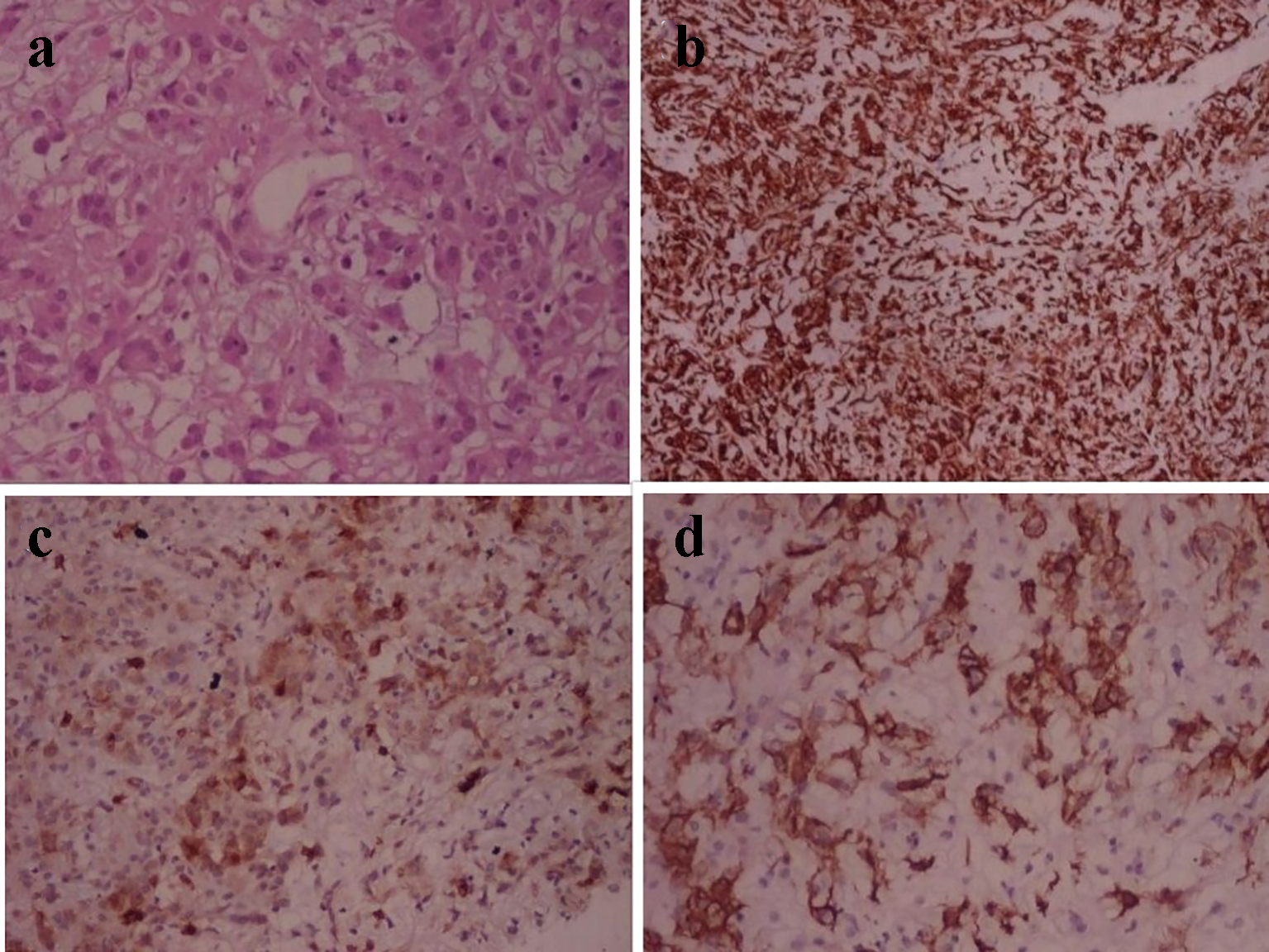 Click for large image | Figure 2. Photomicrograph of chordoid glioma. (a) H&E stain shows cord like arrangement of epithelioid cells in myxomatous background. (b) Immunohistochemistry shows diffuse GFAP positivity. (c) EMA marks plasma cells. (d) CD34 shows patchy positivity. |
On follow-up patient developed two episodes of generalized seizure. CT scan revealed remnant mass and hydrocephalus. Ventriculoperitoneal shunting was performed for obstructive hydrocephalus. Follow-up MRI after 2.5 months revealed 3.4 × 3.5 × 3.8 cm mass, hypointense on T1W and hyperintense on T2W and FLAIR images.
| Discussion | ▴Top |
Chordoid glioma is a rare central nervous system neoplasm considered to be indolent, but still carries an increased risk of recurrence in cases of sub-optimal resection. They are classified as grade II tumors by WHO and the current mainstay of treatment consists of surgical intervention with the possibility of postoperative radiotherapy in cases of subtotal resection (STR).
A total of 81 patients have been reported till 2015. Due to characteristic location within third ventricle, nearly all patients presented symptomatically. Only three of 81 (3.7%) patients presented incidentally. Among the remaining 78 patients, the most common presenting symptoms, in order, were headache, visual symptoms, mental status changes and memory deficits, nausea/vomiting, and lethargy/somnolence. Less frequently reported symptoms included ataxia/gait instability, seizures, syncopal episodes, and speech difficulties. History of endocrine dysfunction was noted in some patients, including amenorrhea in four, diabetes insipidus in four, and hypothyroidism in two patients. Psychotic features were noted in two patients [3]. Primary polydipsia as a presentation of chordoid glioma has not been reported previously. One case of hyponatremia due to SIADH in chordoid glioma has also been reported [5].
The sustained increase in water intake in patients with primary polydipsia may be due to hypothalamic lesions of thirst center (tumors, infiltrative disease) or more commonly due to underlying psychiatric illness. Differentiation of primary polydipsia from diabetes insipidus as a cause of polyuria and polydipsia is important to avoid water overload, which can result from inappropriate vasopressin therapy. The water deprivation test is the gold standard test to differentiate central or nephrogenic diabetes insipidus from primary polydipsia in patients with polyuria and polydipsia. A threshold for urine osmolality of > 800 mOsm/kg after water deprivation yielded a sensitivity and specificity of 96% and 100%, respectively, for diagnosing primary polydipsia. Sensitivity increases to 100% if cut-off value for urine osmolality was set at 680 mOsm/kg [6].
Chordoid gliomas are thought of as being well encapsulated and non-invasive. Al Hinai and Petrecca reported the only case of chordoid glioma that involves the optic chiasma [7]. Our case also has involvement of optic chiasma.
Chordoid glioma is hyperdense on CT in 65% of cases, and enhances homogenously following contrast administration in 68%. Chordoid glioma may be confused with other hyperdense lesions such as lymphoma, meningioma and aneurysms. MRI is preferred for chordoid glioma. Typically these tumors are well circumscribed, round or oval in shape on T1W MR (63%) of cases and slightly hyperintense on T2W images, with homogeneous enhancement by gadolinium (70%). Most tumors are solid, but cystic changes are infrequently seen within the periphery of these tumors [8].
Histopathologically, the tumor consists of clusters and cords of oval to polygonal epitheliod-shaped cells with eosinophilic cytoplasm, which are embedded in a mucinous matrix. Mitoses are rare and necrosis is absent. Lymphoplasmacytic infiltrate and Russell bodies are often found in the tumor. Immunohistochemically, chordoid glioma is strongly and diffusely positive for GFAP and vimentin, and shows focal reactivity to EMA and cytokeratin in some cases, but little or no reactivity to S-100 protein. Some authors reported immunoreactivity for CD34, which is helpful for making differential diagnosis between chordoid meningioma, pilocytic astrocytoma, and/or ependymoma. The histogenesis of chordoid glioma remains uncertain. The immunohistochemical positivity for GFAP and vimentin favors glial origin [9].
There is controversy over the best treatment for chordoid glioma. Most reported cases were surgically resected, either gross total or subtotal resection, while other had stereotactic biopsy. The current treatment of choice for chordoid glioma is surgical resection. Gross total resection (GTR) is the ideal treatment, although tumor size and location often impede radical resection. Radiotherapy has largely been reserved as adjunctive therapy for management of residual tumor following resection [3, 8].
Mortality rates and complications related to surgical procedures vary according to surgical technique. GTR has led to postoperative death in 29% of cases, and 67% chances of complications among survivors. After partial resection, mortality and morbidity decreased to 14% and 50%, respectively. The most common postoperative complication reported are cognitive disorders, pulmonary embolism and hypothalamic dysfunction (DI, SIADH, weight gain, hypothyroidism and panhypopituitarism) [3, 8]. The rate of 1-, 3- and 5-year progression free survival (PFS) was 88.4%, 82.5%, and 70.7%, respectively. The extent of tumor resection was the most robust predictor of improved rate of tumor control [3].
Jung and Jung have reported third ventricular chordoid glioma with unusual aggressive behavior and recurrence in 3 months which is the shortest duration before recurrence. Our patient experienced recurrent lesion after 2.5 months [10].
Conclusion
Most physicians have little knowledge of this relatively new central nervous system tumor. Based on this new case presentation with aggressive behavior in some patients, diagnosis of chordoid glioma should be kept in mind in dealing with patients of primary polydipsia. GTR should be the primary goal for the management of chordoid glioma whenever possible, as it is associated with improved rates of tumor control without increased rate of postoperative complications.
Conflicts of Interest
There are no conflicts of interest involving the authors and contents in this manuscript.
Financial Support
None.
Author Contributions
All authors have contributed to manuscript conception, design, and drafting. Final approval of the version is given by all authors.
| References | ▴Top |
- Brat DJ, Scheithauer BW, Staugaitis SM, Cortez SC, Brecher K, Burger PC. Third ventricular chordoid glioma: a distinct clinicopathologic entity. J Neuropathol Exp Neurol. 1998;57(3):283-290.
doi pubmed - Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114(2):97-109.
doi pubmed - Ampie L, Choy W, Lamano JB, Kesavabhotla K, Mao Q, Parsa AT, Bloch O. Prognostic factors for recurrence and complications in the surgical management of primary chordoid gliomas: A systematic review of literature. Clin Neurol Neurosurg. 2015;138:129-136.
doi pubmed - Pomper MG, Passe TJ, Burger PC, Scheithauer BW, Brat DJ. Chordoid glioma: a neoplasm unique to the hypothalamus and anterior third ventricle. AJNR Am J Neuroradiol. 2001;22(3):464-469.
pubmed - Calanchini M, Cudlip S, Hofer M, Byrne J, Fabbri A, Grossman A. Chordoid glioma of the third ventricle: a patient presenting with SIADH and a review of this rare tumor. Pituitary. 2016;15:1-6.
doi - de Fost M, Oussaada SM, Endert E, Linthorst GE, Serlie MJ, Soeters MR, DeVries JH, et al. The water deprivation test and a potential role for the arginine vasopressin precursor copeptin to differentiate diabetes insipidus from primary polydipsia. Endocr Connect. 2015;4(2):86-91.
doi pubmed - Al Hinai QS, Petrecca K. Rarest of the rare: Chordoid glioma infiltrating the optic chiasm. Surg Neurol Int. 2011;2:53.
doi pubmed - Morais BA, Menendez DF, Medeiros RS, Teixeira MJ, Lepski GA. Chordoid glioma: Case report and review of the literature. Int J Surg Case Rep. 2015;7C:168-171.
doi pubmed - Kobayashi T, Tsugawa T, Hashizume C, Arita N, Hatano H, Iwami K, Nakazato Y, et al. Therapeutic approach to chordoid glioma of the third ventricle. Neurol Med Chir (Tokyo). 2013;53(4):249-255.
doi - Jung TY, Jung S. Third ventricular chordoid glioma with unusual aggressive behavior. Neurol Med Chir (Tokyo). 2006;46(12):605-608.
doi
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Neurology Research is published by Elmer Press Inc.