
| Journal of Neurology Research, ISSN 1923-2845 print, 1923-2853 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Neurol Res and Elmer Press Inc |
| Journal website https://www.neurores.org |
Review
Volume 10, Number 5, October 2020, pages 150-159
The Effect of Edaravone on Amyotrophic Lateral Sclerosis
Brandon Nightingale
Saba University School of Medicine, Church Street, The Bottom, Saba, Dutch Caribbean
Manuscript submitted July 6, 2020, accepted July 18, 2020, published online August 12, 2020
Short title: Effect of Edaravone on ALS
doi: https://doi.org/10.14740/jnr589
| Abstract | ▴Top |
Amyotrophic lateral sclerosis (ALS), a neurodegenerative disease, is fatal within 3 years of symptom onset. Both upper motor neurons and lower motor neurons are targeted. It is hypothesized that: edaravone is effective at managing ALS. This review article used a combination of secondary and primary research articles to gain a plethora of information to help test this hypothesis. Using PubMed, research articles were studied to identify important information. For the Introduction, both secondary and primary articles were used without a limitation on publication date. For the Results section, only primary articles were used which had to have been published no earlier than 2006. The Results section of this review helped to support the hypothesis that edaravone is effective at managing ALS. The most pivotal efficacy endpoint, the change in the Amyotrophic Lateral Sclerosis Functional Rating Scale Revised score, was positively influenced by edaravone over the placebo. This was shown to be statistically significant by use of analysis of variance, amongst other statistical tests. Secondary endpoints such as forced vital capacity and pinch strength were also analyzed, showing similar favorable results. From the clinical trials analyzed in this review, it is concluded that edaravone is sufficient in treating ALS. Edaravone is limited to a target population which could prove to be a problem. Future studies should explore this issue in hopes of expanding the treatment population of edaravone.
Keywords: Amyotrophic lateral sclerosis; Neurodegenerative disease; Edaravone; ALSFRS-R; Forced vital capacity; Pinch strength; Clinical trials
| Introduction | ▴Top |
Amyotrophic lateral sclerosis (ALS), more informally known as Lou Gehrig’s disease, is an adult-onset, severely debilitating neurodegenerative disease that has a steady and progressive course. Once thought to be a pure motor neuron disease, ALS is now widely accepted to be a multisystem disease involving other domains such as cognition and behavior [1]. The disease is known as a heterogeneous disorder, meaning that the same disorder can be caused by myriad gene mutations [2]. ALS is associated with ≥ 30 gene mutations, with evidence of oligogenic inheritance and genetic pleiotropy [3]. This is different from a homogeneous disorder in which the catalytic event for the disorder is the same for all patients. ALS is considered either sporadic (SALS) or familial (FALS), in which 10% of the cases are inherited in a familial pattern [4].
The characteristic feature for ALS is the involvement of both upper motor neurons (UMNs) and lower motor neurons (LMNs), which present with defining features when a lesion occurs in the respective neuron. When a lesion takes place in an LMN, the characteristic findings will be weakness, muscle atrophy and fasciculations. In contrast, when a lesion takes place in an UMN, the characteristic findings will be hyperreflexia and hypertonia [2]. ALS has variable presentation from patient to patient; however, ALS begins with arm and leg weakness in two-thirds of patients, and the first symptoms are usually asymmetrical and include foot drop, problems with walking, decreased hand dexterity and weakness when lifting the arms [5]. The phenotypical variabilities associated with ALS will be mentioned in more detail below.
There are no geographical limitations on the occurrence of ALS; however, the incidence is varied across different regions and ethnicities [1]. The cause for this difference is most likely due to there being many genes involved in the pathogenesis of ALS, and the different environmental risk factors that present in varying locations. Different risk factors will be discussed in more detail in the following sections.
Pathogenesis
The pathognomonic feature of ALS is the involvement of both UMN and LMN. The initial insult leading to the motor neuron dysfunction is still not yet fully understood. It is known that the neuropathological hallmark of the disease is the aggregation of ubiquitylated proteinaceous inclusions in motor neurons. However, researchers have speculated that the aggregations themselves may not be the toxic substrate, but that the high-molecular-weight complexes (HMWCs) that precede the aggregates are the toxic species [3]. The shedding of these HMWCs acts in a prion-like manner, leading to the propagation of the disease [6].
ALS is a multisystem disease consisting of an array of mutations and several different phenotypical variants. It is severely debilitating, having an overall life expectancy of 3 years, with respiratory failure being the most common cause of death [5]. The vast amount of mutations makes it hard to understand the pathophysiology in full, but recent research has proven to be helpful in this feat.
One of the more commonly researched genes containing mutations relevant to ALS is the superoxide dismutase gene (SOD1), which is essential in reducing oxidative stress on the cell through the scavenging of oxygen radicals. Studies have demonstrated that the mutation in the SOD1 gene is a gain-of-function mutation leading to the accumulation of more reactive oxygen species (ROS). These ROS are highly toxic to the cell as they react with many organic molecules facilitating damage [7]. However, it is almost impossible to measure the levels of oxidative stress directly, so instead researchers measure the levels of oxidative stress biomarkers. These biomarkers are species such as products of DNA oxidation (8-hydroxy-2’-deoxyguanosine), lipid oxidation (4-hydroxy-2,3-nonenal), or malondialdehyde (MDA), and protein oxidation (3-nitrotryosine (3NT)) [8]. The research performed on mutations of SOD1 has led to oxidative stress being a target for pharmaceuticals such as edaravone (Radicava™).
Phenotypic variability
To diagnose ALS, there must be evidence of involvement of both UMN and LMN. Thus, a spectrum exists which patients fall upon, where on one end they have primarily UMN signs, and on the other end they have primarily LMN signs. When there is LMN dominance, there is a question as to whether it is ALS or progressive muscular atrophy (PMA), which resembles the characteristics of ALS. Both ALS and PMA are asymmetric in their presentation, with the only difference being that PMA lacks obvious UMN signs. There is evidence however that points towards PMA simply representing the end spectrum of ALS where LMN signs dominate. For instance, certain SOD1 mutations are associated solely with LMN symptoms, and there are imaging studies that display widespread frontal lobe abnormalities in PMA that are also present in ALS [2]. The other supporting evidence for this theory is that, on autopsy of patients with isolated LMN signs, there is evidence of involvement of the lateral spinal cord (UMN) [9]. Signs of an LMN injury, as mentioned above, are weakness, atrophy and fasciculations in the muscle.
On the opposite end of the spectrum, where UMN signs dominate, which entail hyperreflexia and hypertonia, is where many researchers believe primary lateral sclerosis (PLS) fits. PLS is a neurodegenerative disorder that is represented by isolated UMN signs. Therefore, it has long been thought to be its own separate disorder and not a variant of ALS. The diagnosis for PLS is difficult because many patients will end up developing LMN signs later in the course of the disease. Thus, the diagnosis of PLS must be made after substantial time has passed [2]. Evidence supporting that PLS is in fact a bookend on the spectrum of ALS is seen in autopsies of patients with PLS where there is obvious LMN involvement [10].
Bulbar-onset ALS (BALS), which happens in around 20% of patients, is where weakness starts in the bulbar muscles (jaw, face, soft palate, pharynx, larynx and tongue). Common symptoms associated with BALS are dysarthria (93% of patients), dysphagia (86% of patients), and tongue fasciculations (64% of patients) [11]. There is also a phenomenon known as pseudobulbar affect which is uncontrolled laughing or crying [2]. This variant of ALS is known as a very aggressive form and is associated with a poorer prognosis than the spinal onset variant. This is often due to the fact that patients with BALS are prone to aspiration and nutritional problems due to involvement of bulbar muscles. They are also more prone to respiratory problems which contributes to early death [12].
Genetics
As stated above, there are many genes that are associated with the pathogenesis of ALS, but around two-thirds of all familial cases are thought to involve only four genes, Chromosome 9 Open Reading Frame 72 (C9ORF72), SOD1, Fused in Sarcoma (FUS), and TAR DNA Binding Protein (TARDBP) [13]. Of these four genes, the first to be discovered was SOD1. The SOD1 gene mutation accounts for up to 20% of all FALS cases and is associated with an autosomal dominant inheritance pattern [5]. This gene is found on chromosome 21 and is associated with ≥ 160 mutations; thus, there is vast phenotypic heterogeneity. For example, the A4V mutation, most common in North America, presents as a highly aggressive form of ALS which typically results in death within a year of the first symptoms. On the other hand, the homozygous D90A mutation has a much more torpid course, seeing the patient develop respiratory problems far later in the disease [4]. The vast amount of mutations associated with a single gene make it difficult to understand the pathogenicity of each specific mutation and make it challenging to map out its penetrance [13]. However, what makes mutations in SOD1 so unique is that they do not incorporate the pathology associated with TDP-43 and/or FUS which is found in almost every other case [4].
The TARDBP mutation accounts for roughly 4% of FALS cases, and an even smaller percentage of SALS. The discovery of this mutation was of great significance as the TARDBP gene encodes for TDP-43, the major component in ubiquitin positive neuronal inclusions. These inclusions are the pathological hallmark of ALS and fronto-temporal dementia (FTD) [4]. TARDBP is thought to be associated with about 50 mutations which lead to TDP-43 positive inclusions in the brain. These inclusions will go on to alter the processing of RNA [5].
The FUS gene is associated with the cause of chromosome 16p-linked FALS, and accounts for roughly 4% of FALS, and less than 1% of SALS [4]. All but one of the mutations associated with FUS is inherited in an autosomal dominant pattern, and the gene mutation is pathognomonic for FUS-positive TDP-negative inclusions [5]. These inclusions are special in nature because they are absent of ubiquitin positive and TDP-43 positive aggregates. One theory as to why these inclusions contain no TDP-43 aggregates is that FUS acts downstream of TDP-43 [4].
The C9ORF72 gene accounts for most of the FALS cases (40%) and to a lesser extent some of the SALS cases (7%). There is a large hexanucleotide repeat expansion in the gene (GGGGCC) which is the cause of chromosome 9p21-linked ALS and FTD. The repeat expansions alter RNA metabolism which causes damage to motor and frontal cortex neurons [4]. As was the case with the TARDBP mutation, there is accumulation of TDP-43 protein in the brain [5].
Epidemiology
Recent epidemiological research has shed light on the disparity in the incidence of ALS across different regions of the world, and amongst different ethnicities. ALS does not specifically target any sub-population; however, it is known that the disease has a higher prevalence in Caucasians, men, non-Hispanics, people over the age of 60, and persons with a history of FALS [14]. A recent study demonstrating statistics from 2015 found the prevalence of ALS in the USA to be 5.2 per 100,000, with a total 16,585 cases. This same study also found the common theme of which sub-populations have a higher prevalence of ALS to hold true, where the lowest prevalence was in persons aged 18 - 39 (the highest being in persons aged over 60), men have a much greater prevalence than women, and white people have a greater prevalence than black people [15].
In Europe, a recent study found the incidence of ALS to be 2.2 per 100,000 person-years for the general population. Areas such as East Asia (0.89 per 100,000 person years) and South Asia (0.79 per 100,000 person years) have the lowest incidence levels. The wide margin of difference is most likely due to the many different genes that are in play and the potential for different environmental risk factors [1]. The environmental risk factors are not fully known, but some research has been done to show that exposure to smoking, lead, heavy metals and electromagnetic fields may increase the risk of ALS, whereas type 2 diabetes mellitus, female contraceptive hormones and high lipid levels may decrease the risk of developing ALS [3, 16].
Survival is also different based on location. In Europe survival is much shorter than that in Central Asia (24 months and 48 months respectively). There is also a difference in survival time when comparing people of mixed ancestry with those who are not. In Cuba, when comparing the population of mixed ancestry versus the population of white or black individuals the mortality rates were of significant difference [3]. Understanding the epidemiology and mortality of ALS is important in creating a clinical profile for patients, and as more research on this topic is done perhaps a better individualized care plan can be created.
Treatment
There are currently two FDA approved drugs for the treatment of ALS, riluzole and edaravone. Riluzole, the first FDA approved drug for ALS, has been shown to increase survival by 3 months, and has no substantial effect on muscle improvement. The mechanism of action of riluzole is not fully understood; however, it is believed that the drug works by blocking voltage gated sodium channels which reduces glutamatergic neuron transmission [3].
Edaravone, also known as MCI-186, the most recent FDA approved drug for ALS (2017), has shown promising results in selective patients. The drug is known to be a free radical scavenger, eliminating oxidative stress and cellular damage by removing lipid peroxides and hydroxyl radicals [17]. These measures are in part due to the transferring of an electron, from the drug, to the radical [18]. Through the scavenging of free radicals, edaravone has shown protective effects on neurons, glia, and vascular endothelial cells and has demonstrated the ability to decrease the inflammatory response of activated microglial cells [8].
Rationale
This review will explore several recent clinical trials demonstrating the effects of edaravone on ALS patients. With an understanding that oxidative stress is a major factor in neuron damage, it is proposed that a free radical scavenger can have a substantial impact on the pathogenesis of ALS [18]. With proven success in a specific sub-population of ALS patients, edaravone has the potential to alter the current course of ALS and change the way clinicians manage their patients.
| Literature Search | ▴Top |
For this review article, several topics were researched in-depth with the help of PubMed. The Introduction of this article was designed to gain an understanding of ALS from its symptom presentation to its epidemiological pattern. For this, there was no limit on specific dates on which an article had to be published. This is because there are important articles that were published many years ago that include background information that is key to understanding ALS. There was also no limitation on whether an article was a primary or a secondary article. Search criteria that were used to obtain these articles were: ALS, FALS, ALS treatment, ALS epidemiology, ALS presentation, ALS phenotypical variants, ALS genetics, edaravone, free radical scavenger, oxidative stress, ROS, neuron damage, neurodegeneration, superoxide dismutase, and superoxide dismutase gain of function mutation in ALS.
For the Results section of this paper, only primary research articles were used (Table 1). The use of primary articles, focused around clinical trials, helps to hone in on the hypothesis of this review article, in regards to the use of edaravone for ALS. Inclusion criteria were that the papers had to be primary, and they could not be published before 2006 (Fig. 1). I chose to limit the publication date to 2006 as to make sure the information on the drug of interest was as recent as possible. The search criteria that were used to obtain these primary articles were very similar to those used in the Introduction. In addition to the search terms used above, the therapeutic effects of edaravone, edaravone as a free radical scavenger, edaravone for ALS, and edaravone as a protective measure against neurodegeneration were used. Most of the primary articles are from the same journal. This is because edaravone is a relatively new drug, especially in relation to the treatment of ALS; thus, there are not an abundance of clinical trials yet. Also, many of the clinical trials are post hoc analyses, or extension studies, on previous trials, in which the researchers are searching for specific sub-populations or safety measures (Supplementary Material 1, www.neurores.org).
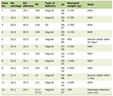 Click to view | Table 1. Study Design of Final Selected Publications |
 Click for large image | Figure 1. Flowchart illustrating selection of primary articles. |
| Literature Review | ▴Top |
An open trial study, published in 2006, looked at the potential therapeutic effects of edaravone in treating ALS. Specifically, this study was designed to determine if there was a connection between the free radical scavenging capabilities of edaravone and the known oxidative stress present in the pathogenesis of ALS. It is known that 3NT is a marker of oxidative cellular damage, and that this biomarker is elevated in post-mortem examination of patients with ALS. Also, oxidative lesions are often present in nervous tissue of patients with ALS. Therefore, there is a real potential for exploring a free radical scavenger and its ability to treat ALS [18].
The inclusion criterion for the study was that there had to be a diagnosis of either SALS or FALS. As for the exclusion criteria, it was as follows: there could not be a tracheotomy, artificial respiration, or dyspnea, no advanced cancer, no severe cardiac insufficiency, stable ALS Functional Rating Scale Revised (ALSFRS-R) score, and no person under the age of 20 years [18]. A person meeting both the inclusion and exclusion criteria, after giving informed consent, could participate in the study.
The protocol for administration of edaravone and the observation periods will be listed below. Either 30 or 60 mg of edaravone, dissolved in 100 mL of saline, was given to patients once per day via an intravenous (IV) drip. This was done for 14 consecutive days which was then followed by a 2-week observational period. The observation period was set in place to check for any side effects that may result due to edaravone. If there were no side effects noted, then edaravone would again be administered once a day for 2 weeks followed by a 2-week observational period. This process was repeated five times, giving the study a total length of 6 months [18].
Since 3NT is a potent biomarker of oxidative damage, an enzyme immunoassay (EIA) was used to measure the levels of 3NT in cerebrospinal fluid (CSF). The primary endpoint used for this study was the change in ALSFRS-R score 6 months after completion of the edaravone administration period. There were also secondary endpoints that were measured which included muscle function, respiratory function, blood gases, CSF protein, 3NT, and lipid peroxide levels in the CSF and blood [18]. These secondary endpoints were also assessed 6 months following the completion of the edaravone administration period.
This study used the Wilcoxon signed rank test to determine the statistical significance of differences in rates of decline of ALSFRS-R score, and the criterion of statistical significance was set at P < 0.05. At the beginning of the study there were initially 20 participants, of which four patients ended up exiting the study early and did not get factored into the data [18]. This left a total of four subjects in the 30-mg group, and 12 subjects in the 60-mg group. To compare the changes in the ALSFRS-R score in the 6 months before starting treatment, and the 6 months during treatment, efficacy was evaluated in all 12 subjects in the 60-mg group who completed the entire six treatment cycles. Safety was evaluated for all the subjects [18].
For the results, the study demonstrated that during the 6-month treatment period the decline in the ALSFRS-R score was 2.3 ± 3.6 points, which was significantly less than that in the 6 months prior to treatment with edaravone (4.7 ± 2.1 points) [18]. These data are important as it shows that treatment with edaravone, in the 60-mg group, reduced the rate of decline of the ALSFRS-R score by 2.4 ± 3.5 points (Wilcoxon signed rank test, P = 0.039) [18]. Another interesting, and highly important, discovery was that in almost every patient in the 60-mg group, the levels of CSF 3NT were markedly reduced. The combination of the reduced ALSFRS-R score and the fact that the 3NT levels were reduced indicates that edaravone is effective at treating ALS and reducing oxidative damage.
Outside of 3NT, there are other biological markers that can be used to measure the levels of oxidative damage. An open trial study measured the amount of oxidative damage, and the effect of edaravone on these measurements, by looking at the levels of the redox balance of plasma coenzyme Q10, plasma uric acid (UA), and the percentage of polyunsaturated fatty acids (PUFAs) in total plasma free fatty acids (FFAs). The study consisted of 26 participants that had to have a diagnosis of SALS or FALS. The exclusion criteria were that no subject could have compromised respiratory function, any cancers or severe cardiac insufficiency, or be less than the age of 20 [19].
The study drug, edaravone, was dissolved in 100 mL of saline and then administered intravenously at a rate of one to four times a week. Amongst the 26 subjects, 17 received edaravone for at least 3 months, and the other 13 subjects continued the treatment for 6 months. Researchers collected heparinized blood from the subjects before and after the administration of edaravone at week 0, week 1, month 1, month 3, and month 6. The ALSFRS-R score was also measured at the same interval periods during this study [19].
As for the results and data for the study, the statistical significance of differences was calculated with Student t-test, and to measure the time course of efficacy of edaravone administration the post-treatment data were assessed using repeated-measures analysis of variance (ANOVA). The group differences were analyzed with Fisher’s exact probability test, and the P value, determining what is statistically significant, was set at P < 0.05 [19].
Results showed that there was no significant difference in plasma antioxidants and total coenzyme Q10 between the 26 study subjects and a control group consisting of 55 age-matched healthy controls. But there was a significant decrease in UA and plasma bilirubin (BR). The change in ALSFRS-R score in the 26 subjects showed a significant improvement at the end of the 6 months (P < 0.008). This study showed that edaravone is effective at slowing down the progression of ALS; however, it does indicate that edaravone alone is not effective in reducing coenzyme Q10 levels [19].
Due to the displayed success of edaravone in treating ALS and oxidative damage in previous studies, as listed above, edaravone has become a key player in clinical trials geared towards ALS. To date, there are four different ALS study groups that have carried out different clinical trials: The Edaravone (MCI-186) ALS 16 Study Group, The Edaravone (MCI-186) ALS 17 Study Group, The Edaravone (MCI-186) ALS 18 Study Group, and The Edaravone (MCI-186) ALS 19 Study Group. In the following paragraphs a more in-depth analysis of these study groups will be shown, with the goal being that a better picture can be drawn with regards to edaravone and its clinical impact on ALS.
In 2014, a randomized control trial (MCI186-16), which was double blinded and featured a placebo group, aimed to study the efficacy and safety of edaravone with respect to treating ALS. This study was the first phase III clinical study of edaravone for ALS. The inclusion criteria for this study were as follows: subjects’ age had to fall between 20 and 75 years, they had to have a diagnosis of either definite, probable or probable laboratory-supported ALS, they had to have a forced vital capacity (FVC) of at least 70%, the duration of disease had to be within 3 years, the change in ALSFRS-R score during the 12-week pre-observational period had to be between -1 and -4 points, and the subjects had a Japanese ALS severity classification of 1 or 2. Having a Japanese ALS severity classification of 1 or 2 meant that the patients were either able to work or perform housework (1) or that they were independent in their living, but were unable to work (2) [17]. This study also had exclusion criteria which were as follows: reduced respiratory function and complaints of dyspnea, any disorder that would complicate evaluating the efficacy of edaravone, any complication requiring hospitalization, any infections that require antibiotics, renal dysfunction marked by creatinine clearance of 50 mL/min or less within 28 days before treatment, and any undergoing cancer treatment [17].
Before any randomization happened, which placed patients either in the treatment group (receiving edaravone) or in the placebo group (receiving saline), there was a 12-week pre-observation period. During the randomization period researchers used dynamic allocation, which involves the use of non-random algorithms, to minimize the effects of three factors: change in ALSFRS-R score during the pre-observation period (-4, -3 or -2, -1), disease presentation (bulbar or limb), and use of riluzole (yes or no) [17].
The study period was 9 months, which consisted of the 12-week pre-observational period followed by a 24-week treatment period. In the treatment group, edaravone was administered in 60 mg doses intravenously for 1 h once per day. In the placebo group, saline was administered following the same dosing guidelines in the treatment group. One treatment cycle was composed of 2 weeks of study drug administration followed by a 2-week observational period. During the first cycle, edaravone was given every single day during the treatment period, and for each cycle thereafter (cycles 2 - 6) edaravone was given for 10 out of the 14 days during the treatment period [17].
The primary efficacy endpoint of this study was the change in the ALSFRS-R score. There were also several other secondary endpoints [17]. To evaluate these changes, patients were analyzed before the pre-observation, before the start of treatment cycle 1, and at the end of each treatment cycle. This allowed for a detailed analysis of the changes that took place throughout the study.
The study aimed for a target population of 200 patients. The statistical power of the study was set at 85% when at least 100 patients were enrolled per group. To measure the change in ALSFRS-R scores during treatment, which is the difference between the score before the start of the first treatment cycle and the score at 2 weeks after the end of the last treatment cycle, an analysis of covariance (ANCOVA) was used. The efficacy of edaravone could only be noted if a significant inter-group difference was found. To interpret the main effect of the study, a two-sided level of significance of 5% and a two-sided 95% confidence interval (CI) were chosen, and to determine the existence of effect on interaction a two-sided level of significance of 15% was chosen [17]. Researchers used ANCOVA and repeated-measures ANOVA for the secondary endpoints as well.
Results from the study showed that the inter-group difference (changes in the ALSFRS-R scores) was 0.65 points by ANCOVA which was not statistically significant. There was also no statistically significant inter-group difference when repeated-measures ANOVA was used. Pinch strength, one of the secondary endpoints, did show statistical significance in favor of the edaravone group (P = 0.038) [17]. Thus, this study did not demonstrate efficacy of edaravone in halting the progression of ALS.
A post hoc subgroup analysis focusing on the outcomes of MCI186-16 aimed to pinpoint a specific sub-group that edaravone may show efficacy in [20]. As a continuation study of MCI186-16, this study wanted to find the reasons as to why edaravone did not show efficacy in delaying the progression of ALS. The study design for this post hoc analysis was kept the same as the original study design in MCI186-16, making the change in ALSFRS-R score the primary endpoint. To find a sub-group population in which the efficacy of edaravone can be demonstrated there were two sub-group populations created: efficacy-expected sub-population (EESP), and the greater-efficacy-expected sub-population within the EESP (dpEESP2y). Dynamic allocation was also used in this process focusing on three different factors: 1) Change in the ALSFRS-R score during pre-observation; 2) Initial symptom being either bulbar or limb; and 3) Concomitant use of riluzole. Those who were allocated into the EESP group had an FVC of ≥ 80% before treatment and ≥ 2 points for all item scores in the ALSFRS-R before treatment. Those who were allocated into the dpEESP2y group had a diagnosis of definite or probable ALS and were within 2 years of initial ALS symptom onset at the time of giving informed consent [20].
To analyze the data in this study researchers used ANOVA to determine the changes in ALSFRS-R scores during treatment, which consisted of the time frame from before treatment was started to 24 weeks after starting treatment. Researchers also used ANOVA to analyze secondary endpoints which consisted of %FVC, Modified Norris Scale score, and 40 items ALS assessment questionnaire (ALSAQ-40) score [20].
At the end of the study the populations per group were as such: the full analysis set (FAS) consisted of 205 patients (104 patients in the placebo group, and 101 patients in the edaravone group), the EESP group consisted of 104 patients (50 patients in the placebo group, and 54 patients in the edaravone group), and the dpEESP2y group consisted of 72 patients (32 patients in the placebo group, and 40 patients in the edaravone group) [20]. The differences of the least square mean between groups in regards to the change in the ALSFRS-R score ± standard error (SE) during treatment were as follows: 0.65 ± 0.78 (P = 0.4108) in the FAS, 2.20 ± 1.03 (P = 0.0360) in the EESP, and 3.01 ± 1.33 (P = 0.0270) in the dpEESP2y [20]. From this data, it can be seen that there was a significant inter-group difference in the EESP and dpEESP2y; however, there was not a significant inter-group difference in the FAS. Expanding upon this further, it is seen that the inter-group difference was largest for the dpEESP2y. The results for the secondary endpoints maintained the same trend as that of the primary endpoint. The inter-group differences of %FVC and the Modified Norris Scale score were largest for the dpEESP2y [20].
This study proved that edaravone had a substantial impact on inhibiting the progression of functional disorder in patients with ALS in a specific sub-group of patients (EESP and dpEESP2y). In addition to the positive effects of edaravone listed above, this study also found that there were no notable safety issues in either the EESP or the dpEESP2y. Finding a specific sub-population of ALS in which edaravone is effective is a starting point for finding the right niche in which edaravone can be included in treatment plans.
After the completion of MCI186-16 an extension study was performed (MCI186-17) to evaluate the longer-term efficacy and safety of edaravone. Patients were enrolled into this extension study if they completed treatment in the original study without meeting any of the exclusion criteria which included renal failure at the end of cycle 6. A total of 181 patients were enrolled into the extension study out of a possible 183 patients. The FAS consisted of 180 patients. Patients in the original study who had received edaravone were randomized into either the edaravone or placebo group in the extension study, while those who originally were in the placebo group were all assigned to the edaravone group in the extension study. The extension period took place for 6 months (cycles 7 - 12), and after this period all patients could receive edaravone for an additional 3 months (cycles 13 - 15) [21].
The efficacy endpoints for this study were the changes in the ALSFRS-R score, time to death, disease progression, %FVC, Modified Norris Scale score, ALSAQ score, hand grip strength, and pinch strength. To evaluate these endpoints ANOVA was used, and each endpoint was compared between the edaravone-edaravone (E-E) and the edaravone-placebo (E-P) groups from the baseline (before starting cycle 7) to 6 months after the baseline (the end of cycle 12). The level of significance for the FAS in this study was set at 5% (two-sided) [21].
The inter-group difference amongst the E-E and E-P did not show statistical significance in either the FAS or the EESP; however, the difference in the EESP (1.85 ± 1.14, P = 0.1127) was greater than that in the FAS (1.16 ± 0.93, P = 0.2176) [21]. Neither the E-E or E-P, in the FAS or the EESP, showed any statically significant changes in endpoints from baseline to the end of cycle 12. There were also no statistically significant differences in the rate of adverse events (AEs) between the E-E and the E-P groups (P = 0.3625). This study was not performed necessarily to see a decline in a patient’s ALSFRS-R score; however, it was to measure the efficacy and long-term safety of edaravone, which it did successfully. The study helped to establish a timeframe in which edaravone continues to show efficacy (up to 15 cycles) [21].
A post hoc analysis of MCI186-17 was conducted which focused on the dpEESP2y, which consisted of the patients who met dpEESP2y criteria at the beginning of study MCI186-16. This study analyzed the changes in ALSFRS-R scores in the dpEESP2y from week 24, the start of MCI186-17, to week 48 which was the end of MCI186-17. This was also done for the sub-populations that met each criterion used to define the dpEESP2y. To compute the data ANOVA was used which was bounded by treatment group and change in ALSFRS-R score during the pre-observation period as fixed effects [22]. The level of statistical significance was set at 95% (P = 0.05).
As stated previously, MCI186-17 had a total of 181 patients enrolled into the study, with a total of 67 patients in the dpEESP2y group. Within the dpEESP2y group, there were 22 patients in the E-E group, 16 patients in the E-P group, and 29 patients in the P-E group. The least square mean difference, with regards to the changes in ALSFRS-R, between the E-E group and the E-P group was 2.79 ± 1.51 in the dpEESP2y (P = 0.0719) [22]. The between-group difference in ALSFRS-R was greatest in the dpEESP2y in both MCI186-16 and MCI186-17, while both the dpEESP2y and EESP showed a greater difference than the FAS. Thus, both studies conveyed that edaravone treatment had a greater impact on the differences in change in ALSFRS-R than did placebo and provide evidence that edaravone is clinically effective at treating ALS in patients who meet dpEESP2y criteria at baseline [22].
MCI186-18, a randomized control trial that was double blinded and featured a placebo group, also continued off the first phase III confirmatory study (MCI186-16). However, instead of patients only requiring a grade of 1 or 2 on the Japan ALS severity classification scale, as in MCI186-16, they needed to have a grade of 3. A grade of 3 meant that these patients were no longer able to live independently. Along with needing a grade 3 on the Japan ALS severity classification sale, patients were also required to meet the following criteria: age 20 - 75 years, a diagnosis of definite, probable, or probable-laboratory-supported ALS, %FVC of at least 60%, duration of the first symptom within at least 3 years, and change in ALSFRS-R score during the 12-week pre-observation period before the administration of edaravone of -1 to -4 points [23]. Patients were excluded from this study if they met any of the following criteria: reduced respiratory function, dyspnea, and renal dysfunction (creatinine clearance of 50 mL/min or below) within 28 days before treatment. The study design was the same as that of MCI186-16.
The endpoints used for measuring efficacy for this study were changes from baseline to the end of cycle 6 (the last cycle) in the ALSFRS-R score, %FVC, Modified Norris Scale score, ALSAQ, grip strength, pinch strength, and time to death or disease progression. Statistical significance for this study was two sided and was set at 5%. To evaluate the change in ALSFRS-R score for before the start of cycle 1 to the end of cycle 6 ANOVA was performed. Repeated measurement ANCOVA was used to evaluate the ALSFRS-R score from the end of cycle 1 to the end of cycle 6 [23]. The other endpoints in the study were analyzed via ANOVA or repeated-measures ANOVA. The last observation carried forward (LOCF) method was used to assign missing data for patients who completed cycle 3 but had missing data at the end of cycle 6. Fisher’s exact test, with a two-sided level of significance of 5%, was used to calculate the safety of the study [23].
There were 27 patients registered at the beginning of the study, but two patients were excluded from the study after the 12-week pre-observation period. The FAS and the safety analysis set included all 25 patients. The edaravone group had four patients that discontinued treatment, and there were 0 patients in the placebo group that discontinued. Being that there were several discontinuations in the edaravone group, and 0 in the placebo group this created an imbalance between the groups. There were no statistically significant differences with respect to the change in ALSFRS-R score between the two groups (P = 0.835 in the edaravone group and P = 0.945 in the placebo group). As was seen with the change in ALSFRS-R score, there were no statistically significant differences in any of the other endpoints. There was also no significant inter-group difference with respect to AEs (P = 1.000), adverse drug reactions (ADRs) (P = 0.593) or serious AEs (SAEs) (P = 1.000) [23].
Based on the results listed above, the study failed to find any statistically significant inter-group difference changes in the ALSFRS-R score or other endpoints. This could be due in part to the limited population size which does not convey much data. It would be hard to amass a sample size large enough to acquire the data needed to show a trend in edaravone being effective in ALS patients with a Japan ALS severity classification scale of 3.
MCI186-19, a phase III randomized control trial, which was double blinded and featured a placebo group looked to define the safety and efficacy of edaravone in patients with an early stage of ALS and meeting certain criteria which will be described below. Patients had to be within 20 - 75 years, have an ALS of grade 1 - 2 in the Japan ALS severity classification, at least a score of two points on each of the 12 items in the ALSFRS-R, %FVC of at least 80%, a diagnosis of definite or probable ALS, a disease duration of 2 years or less, and a decrease of 1 - 4 points in the ALSFRS-R score during the 3-month observation period [24]. The study design is very similar to the previous studies, but a brief recap will be mentioned below. Patients were randomized into either the treatment group (IV edaravone) or the placebo group (IV saline). Edaravone or saline was administered in 60 mg doses for a total of six cycles with each cycle lasting a total of 4 weeks, giving the total duration of the study a time frame of 24 weeks. Cycle 1 saw either edaravone or saline be administered once per day for 14 straight days which was then followed by a 14-day observation period. Cycles 2 - 6 saw that patients received treatment for only 10 days out 14, which was then followed by a 14-day observation period.
The main efficacy outcome of the study was the change in ALSFRS-R score from the beginning of the study to the end of the study (baseline - 24 weeks). The data were compared between the two groups using ANOVA, with statistical significance being set at 0.05 [24]. For those patients who had missing data at the end of cycle 6, a linear regression was used to impute the data while considering the progression of ALS as a factor. Secondary endpoints were also analyzed via ANOVA; however, there was no adjustment for multiplicity. Least square mean differences between the two groups were estimated for the efficacy endpoints (from baseline to the end of cycle 6). A Kaplan-Meier plot, log-rank test and generalized Wilcoxon test were used to analyze the secondary endpoint of time to death or disease progression [24].
Of the efficacy endpoints analyzed by least square mean, there were only three statistically significant values: ALSFRS-R score for which the edaravone group had a value of -5.01 ± 0.64 and the placebo group had a value of -7.50 ± 0.66 (P = 0.0013), Modified Norris Scale scores total for which the edaravone group had a value of -15.91 ± 1.97 and the placebo group had a value of -20.80 ± 2.06 (P = 0.0393), and ALSAQ-40 score for which the edaravone group had a value of 17.25 ± 3.39 and the placebo group had a value of 26.04 ± 3.53 (P = 0.0309). At the end of cycle 6, the mean ALSFRS-R score for those taking edaravone was 37.5 with a standard deviation (SD) of 5.3. This score was similar to the mean ALSFRS-R score for those patients receiving placebo at the end of cycle 4 (37.8 ± 4.1). For the entire study, the difference in the ALSFRS-R score between the two groups was 33% [24]. These results support that edaravone is effective at treating ALS.
A post hoc analysis of MCI186-19 was performed to confirm the statistical robustness of the result. This study used three different statistical methods to analyze the data: 1) All patients were included in the results regardless of treatment cycles received (ALL LOCF) via an ANOVA; 2) A mixed model for repeated measurements (MMRMs) analysis; and 3) The Combined Assessment of Function and Survival (CAFS) endpoint. There was an overwhelming amount of support for edaravone being more superior than the placebo in halting functional decline in ALS [25].
When using ANOVA with ALL LOCF, the difference in the least square mean with SE between the two groups was 2.37 ± 0.75 (P = 0.0019), and when using MMRM it was 2.81 ± 0.78 (P = 0.0004). When using the CAFS endpoint the statistical difference between the two groups was 41.64 ± 12.30 (P = 0.0009). With regards to the distributions of changes in ALSFRS-R total score, using ALL LOCF, from the beginning of the study to the end of the study, there was a shift favoring edaravone over placebo. The edaravone group had 39% of patients showing minimal functional decline (+1, -2 change), while the placebo group had 13% of patients showing minimal functional decline [25]. As stated before, edaravone was also shown to be favored for all 12 items of the ALSFRS-R and for all four domains of the ALSFRS-R. Researchers found that the smallest difference was in the respiratory subdomain, and that the effect in the domains had no preference on either bulbar onset or limb onset [25]. The results of this study show support for edaravone being effective in the management of ALS.
Following the completion of MCI186-19, an extension study was designed to explore the longer-term efficacy and safety of edaravone. This study was offered to all patients who completed the full six cycles in the double-blind period. Out of the possible 137 patients a total of 123 continued to the extension period, with 65 patients being E-E and 58 being P-E [26]. The study featured a discontinuation criterion in which patients had to have a %FVC > 50% and a PaCO2 ≥ 45 mm Hg.
The efficacy endpoints consisted of change in the ALSFRS-R total score, %FVC, Modified Norris Scale score, ALSAQ-40 score, and time to death or disease progression. These endpoints were measured before the pre-observation period, before the start, and end, of the first treatment cycle. As for safety, measurements included the incidence of AE, ADR, SAE, and laboratory tests [26].
The FAS was used to analyze efficacy in the study, while the safety analysis set, which included any patient who received at least one dose of the study drug and had at least one safety data point, was used to analyze safety. A Kaplan-Meier curve was used to analyze time to the first event date of death or disease progression. The change in ALSFRS-R score from baseline (cycle 1) to the end of the extension study (cycle 12) favored those patients who had received edaravone for the entirety of the study: E-E group (-8.0 ± 5.6) versus P-E group (-10.9 ± 6.9). For AE, 81.5% of patients in the E-E group and 82.8% of patients in the P-E group experienced at least one AE, and 26.2% of patients in the E-E group and 39.7% of patients in the P-E group experienced SAE. There was no significant difference in laboratory measurement changes when comparing the two groups. The incidence of ADR in the E-E group was 6.2%, while in the P-E group it was 5.2% [26].
Researchers also found that there was no significant deterioration in the %FVC, Modified Norris Scale, or the ALSQ-40 during this extension period. This, along with the linear change in the ALSFRS-R score for the E-E group throughout the entirety of the study (cycle 1 - 12), and the lack of concern for safety of edaravone, provides no warning that edaravone is un-safe when used for an additional six cycles.
The extension study of MCI186-19 did not use a statistical test to compute their data; thus, a post hoc analysis of this extension study was designed to gather a better interpretation of the efficacy of the change in ALSFRS-R score and other efficacy endpoints. The post hoc analysis focused on an MMRM and the CAFS for assessment, while also analyzing the slopes of time-dependent change between baseline (cycle 1) and the end of cycle 6, and between the end of cycle 6 and the end of cycle 12 using a random coefficient model which included all the available data for each time period [27].
The MMRM, at week 48, showed a decline in ALSFRS-R total score in the E-E group that was significantly less than that in the P-E group (least square mean change from baseline ± SE, 4.17 ± 1.40, P = 0.0037). This finding conferred that the differences in the ALSFRS-R total score during the double-blind period (cycles 1 - 6) were sustained in patients who received edaravone for an additional six cycles. This sustainment was aided by a CAFS endpoint with a P = 0.0089. As for the analysis of the slope, the study found that the double-blind period showed a significant difference between the two groups, while there was not a significant difference between the two groups during the extension period [27]. The above data demonstrates that there is a benefit of administering edaravone early, and continuing the treatment over an extended period, then postponing edaravone treatment. This post hoc analysis displays the beneficial effects of using edaravone in the treatment of ALS, especially when it is used early in the disease process.
A safety analysis for the double-blind period of the three randomized, placebo controlled trials, which are referenced above, found that the overall amount of deaths and discontinuations due to AE were either less in the edaravone group or similar to the placebo group [28]. The total analysis of the study consisted of 368 patients, with 184 in the edaravone group, and 184 in the placebo group. This safety analysis helps to ensure the data extrapolated during the original studies are sufficient in saying edaravone is as safe, if not safer, as the placebo.
| Discussion | ▴Top |
Clinical research directed towards establishing therapeutic evidence for edaravone in treating ALS is centered around 12 clinical trials. While performing these clinical trials, researchers were able to show a connection between the study drug and disease; however, the connection was not always significant. The fact that edaravone has a role in eliminating oxidative stress was consistent throughout all the studies, but the target population of edaravone amongst the studies did vary.
One of the studies found that the reduction in the ALSFRS-R score was not statistically significant between the treatment group and the placebo group, even though the treatment group had a smaller reduction. The thought process then turned towards finding a specific sub-group within the general ALS population in which edaravone would show statistical significance. This sub-group was labeled dpEESP2y. This sub-group was studied further to identify a time-period of effectiveness which was found to be 24 - 48 weeks. Another study also required specific inclusion criteria for patients and this study proved to show efficacy as well. Edaravone only showed efficacy in a defined sub-population, limiting its therapeutic range.
A post hoc analysis also demonstrated results that favored edaravone, showing that the study drug slows functional decline in ALS patients. It was also determined that edaravone should be administered early in the disease cycle and should be continued without interruption. None of the studies demonstrated any increased safety factors for edaravone when compared with the placebo.
There are limitations to these studies which should be factored in when analyzing results. Due to the low number of ALS patients, acquiring a study population of significant numbers of enrollees can often be difficult. This, for obvious reasons, leads to results that are not representative of the entire ALS community. Also, with the varying presentations of ALS (bulbar, limb, familial and sporadic), it is hard to observe, and analyze, changes for the disease as a whole. ALS patients also have a very short life expectancy from the onset of the first symptom, which makes conducting a long-term study challenging.
Currently there are only two FDA approved drugs for the treatment of ALS, riluzole and edaravone. Riluzole is the most widely used drug, partially because it was the first drug approved, but also because edaravone is still a relatively new drug with a limited track record of success. However, riluzole is not highly efficient in extending a patient’s life long-term as it only extends life by an average of 3 months. Therefore, it is highly imperative to find an alternative treatment for ALS, one which inhibits the decline in functionality and extends the patient’s lifespan by more than 3 months. Edaravone has the potential to be that drug. Clinical trials have proven the success of edaravone in slowing the progression of ALS, displaying its abilities to limit the progression of ALS and improve everyday function for patients.
These clinical trials have also shown edaravone to be as safe as a placebo, if not safer, and have shown the ability for edaravone to be carried out for extended periods of time. A pivotal efficacy endpoint used by these clinical trials is the change in ALSFRS-R score between the treatment group and the placebo group. Edaravone proved to be the superior option when analyzing this endpoint. This was also true for many of the secondary endpoints that these trials used such as pinch strength, and %FVC.
Importantly, it should be noted that edaravone has not shown success in every patient with ALS. Currently the greatest success in treatment with edaravone comes in a subset population of patients with ALS, a group labeled dpEESP2y. This group had specific inclusion criteria that needed to be met which can be found above. Thus, the free radical scavenger edaravone should still be assessed clinically to further broaden its therapeutic population, with the hope being that it will eventually be able to help all patients with ALS, and not specifically those who fall into a certain group.
To obtain better results, future studies should aim towards acquiring larger patient populations. This would allow the researchers to have a larger data pool, and the studies would be better positioned for the loss of patients due to death, or meeting exclusion criteria. Future studies should also look to demonstrate the efficacy of edaravone in more sub-populations to broaden the drugs target population. To do this, perhaps different efficacy endpoints could be analyzed that allows new patients to participate in the studies. If clinical trials can obtain a larger patient population, one with a wide array of presentations, then there is a chance that edaravone can show efficacy across the ALS spectrum. In summary, edaravone is effective in treating ALS in several sub-populations, though it is still the most promising therapeutic to date.
| Supplementary Material | ▴Top |
Suppl 1. Evidence Table.
Acknowledgments
I would like to acknowledge Mr. Samuel Johnson and Dr. James Bruzik.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Data Availability
The author declares that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Logroscino G, Piccininni M. Amyotrophic lateral sclerosis descriptive epidemiology: the origin of geographic difference. Neuroepidemiology. 2019;52(1-2):93-103.
- Swinnen B, Robberecht W. The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol. 2014;10(11):661-670.
doi pubmed - Hardiman O, Al-Chalabi A, Chio A, Corr EM, Logroscino G, Robberecht W, Shaw PJ, et al. Amyotrophic lateral sclerosis. Nat Rev Dis Primers. 2017;3:17085.
doi pubmed - Renton AE, Chio A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci. 2014;17(1):17-23.
doi pubmed - Gordon PH. Amyotrophic Lateral Sclerosis: An update for 2013 Clinical Features, Pathophysiology, Management and Therapeutic Trials. Aging Dis. 2013;4(5):295-310.
doi pubmed - Polymenidou M, Cleveland DW. The seeds of neurodegeneration: prion-like spreading in ALS. Cell. 2011;147(3):498-508.
doi pubmed - Mondola P, Damiano S, Sasso A, Santillo M. The Cu, Zn Superoxide Dismutase: Not Only a Dismutase Enzyme. Front Physiol. 2016;7:594.
doi pubmed - Takei K, Watanabe K, Yuki S, Akimoto M, Sakata T, Palumbo J. Edaravone and its clinical development for amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(sup1):5-10.
doi pubmed - Ince PG, Evans J, Knopp M, Forster G, Hamdalla HH, Wharton SB, Shaw PJ. Corticospinal tract degeneration in the progressive muscular atrophy variant of ALS. Neurology. 2003;60(8):1252-1258.
doi pubmed - D'Amico E, Factor-Litvak P, Santella RM, Mitsumoto H. Clinical perspective on oxidative stress in sporadic amyotrophic lateral sclerosis. Free Radic Biol Med. 2013;65:509-527.
doi pubmed - Kuhnlein P, Gdynia HJ, Sperfeld AD, Lindner-Pfleghar B, Ludolph AC, Prosiegel M, Riecker A. Diagnosis and treatment of bulbar symptoms in amyotrophic lateral sclerosis. Nat Clin Pract Neurol. 2008;4(7):366-374.
doi pubmed - Pinto S, Pinto A, De Carvalho M. Do bulbar-onset amyotrophic lateral sclerosis patients have an earlier respiratory involvement than spinal-onset amyotrophic lateral sclerosis patients? Eura Medicophys. 2007;43(4):505-509.
- Chio A, Battistini S, Calvo A, Caponnetto C, Conforti FL, Corbo M, Giannini F, et al. Genetic counselling in ALS: facts, uncertainties and clinical suggestions. J Neurol Neurosurg Psychiatry. 2014;85(5):478-485.
doi pubmed - Mehta P, Kaye W, Raymond J, Wu R, Larson T, Punjani R, Heller D, et al. Prevalence of amyotrophic lateral sclerosis - United States, 2014. MMWR Morb Mortal Wkly Rep. 2018;67(7):216-218.
doi pubmed - Mehta P, Kaye W, Raymond J, Punjani R, Larson T, Cohen J, Muravov O, et al. Prevalence of amyotrophic lateral sclerosis - United States, 2015. MMWR Morb Mortal Wkly Rep. 2018;67(46):1285-1289.
doi pubmed - Oskarsson B, Horton DK, Mitsumoto H. Potential environmental factors in amyotrophic lateral sclerosis. Neurol Clin. 2015;33(4):877-888.
doi pubmed - Abe K, Itoyama Y, Sobue G, Tsuji S, Aoki M, Doyu M, Hamada C, et al. Confirmatory double-blind, parallel-group, placebo-controlled study of efficacy and safety of edaravone (MCI-186) in amyotrophic lateral sclerosis patients. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15(7-8):610-617.
doi pubmed - Yoshino H, Kimura A. Investigation of the therapeutic effects of edaravone, a free radical scavenger, on amyotrophic lateral sclerosis (Phase II study). Amyotroph Lateral Scler. 2006;7(4):241-245.
doi pubmed - Nagase M, Yamamoto Y, Miyazaki Y, Yoshino H. Increased oxidative stress in patients with amyotrophic lateral sclerosis and the effect of edaravone administration. Redox Rep. 2016;21(3):104-112.
doi pubmed - Edaravone Als 16 Study Group. A post-hoc subgroup analysis of outcomes in the first phase III clinical study of edaravone (MCI-186) in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(sup1):11-19.
- Writing Group On Behalf Of The Edaravone Als 17 Study Group. Exploratory double-blind, parallel-group, placebo-controlled extension study of edaravone (MCI-186) in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(sup1):20-31.
- Takahashi F, Takei K, Tsuda K, Palumbo J. Post-hoc analysis of MCI186-17, the extension study to MCI186-16, the confirmatory double-blind, parallel-group, placebo-controlled study of edaravone in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(sup1):32-39.
- Writing Group On Behalf Of The Edaravone Als 18 Study Group. Exploratory double-blind, parallel-group, placebo-controlled study of edaravone (MCI-186) in amyotrophic lateral sclerosis (Japan ALS severity classification: Grade 3, requiring assistance for eating, excretion or ambulation). Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(sup1):40-48.
doi pubmed - Writing G, Edaravone ALSSG. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017;16(7):505-512.
doi - Takei K, Takahashi F, Liu S, Tsuda K, Palumbo J. Post-hoc analysis of randomised, placebo-controlled, double-blind study (MCI186-19) of edaravone (MCI-186) in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(sup1):49-54.
- Writing Group On Behalf Of The Edaravone Als 19 Study Group. Open-label 24-week extension study of edaravone (MCI-186) in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(sup1):55-63.
- Takei K, Tsuda K, Takahashi F, Palumbo J. Post-hoc analysis of open-label extension period of study MCI186-19 in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(sup1):64-70.
doi pubmed - Kalin A, Medina-Paraiso E, Ishizaki K, Kim A, Zhang Y, Saita T, Wasaki M. A safety analysis of edaravone (MCI-186) during the first six cycles (24 weeks) of amyotrophic lateral sclerosis (ALS) therapy from the double-blind period in three randomized, placebo-controlled studies. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(sup1):71-79.
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Neurology Research is published by Elmer Press Inc.