

| Journal of Neurology Research, ISSN 1923-2845 print, 1923-2853 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Neurol Res and Elmer Press Inc |
| Journal website https://www.neurores.org |
Case Report
Volume 14, Number 1, June 2024, pages 43-47

Motor Neuron Disease-Frontotemporal Dementia Spectrum Disorder: A Different Phenotype Related With a Novel TBK1 Gene Variant
Joana Ferreira Pintoa, d, Mariana Santosb, Ana Rita Silvaa, Diana Matosa, c, Vera Fernandesa, Ana Filipa Santosa
aNeurology Department, Hospital de Braga, Braga, Portugal
bNeuroradiology Department, Hospital de Braga, Braga, Portugal
cNeurology Department, Unidade Local de Saude do Alto Minho, Hospital de Santa Luzia, Viana do Castelo, Portugal
dCorresponding Author: Joana Ferreira Pinto, Neurology Department, Hospital de Braga, Braga, Portugal
Manuscript submitted November 4, 2023, accepted March 5, 2024, published online March 14, 2024
Short title: ALS-FTSD With TBK1 Gene Variant
doi: https://doi.org/10.14740/jnr765
| Abstract | ▴Top |
The association of frontotemporal dementia (FTD) and motor neuron disease (MND) is a clinical continuum with genetic and neuropathological overlap. Cohort analysis has been broadening our phenotypic and genotypic knowledge on amyotrophic lateral sclerosis-frontotemporal dementia spectrum disorders. A 57-year-old woman presented with a progressive speech disturbance followed by the development of progressive upper MND in the next 2 years. Rapid progression of disease included anarthria, severe dysphagia requiring a gastrostomy tube and a tetrapyramidal syndrome without lower motor neuron signs. Death occurred within 4 years. Complementary investigation showed a left mesial temporal and frontal lobe atrophy, as well as a corticospinal tract hyperintensity on magnetic resonance imaging (MRI). Hypometabolism in the left parietal-temporal and frontal lobes was also evident in FDG-PET. The association of progressive apraxia of speech with upper motor neuron signs in our patient prompted genetic testing and the finding of a novel TBK1 gene variant. Genetic testing should be considered in patients presenting with FTD, particularly with a positive family history or secondarily associated with an MND. Population-based studies are needed to clarify the prevalence and clinical phenotypes of TBK1 variant carriers.
Keywords: Motor neuron disease; Frontotemporal dementia; TBK1 gene
| Introduction | ▴Top |
The recognition of frontotemporal dysfunction in patients with motor neuron diseases (MNDs) leads to the publication of amyotrophic lateral sclerosis-frontotemporal spectrum disorder (ALS-FTSD) diagnostic criteria in 2009 and its revised version in 2015 [1]. Since then, a growing number of reports have expanded our understanding on the clinical phenotypes, genetic variability and neuropathologic correlations of this disorder.
Frontotemporal dementia (FTD) can present with behavior, personality and/or language abnormalities. The main clinical subtypes include behavioral-variant FTD, semantic dementia and non-fluent primary progressive aphasia (PPA). ALS is characterized by upper and lower motor neuron degeneration resulting in motor weakness, spasticity, hyperreflexia, muscle atrophy and/or fasciculations.
The neuropathological substrate of FTD is most often selective frontal and temporal lobe atrophy, termed frontotemporal lobar degeneration (FTLD). Depending on the specific protein that abnormally aggregates in neurons and glial cells, four major subtypes of FTLD can be recognized. TAR-DNA-binding protein-43 (FTLD-TDP-43) accounts for one of those and is responsible for most cases of ALS-FTSD [2]. Within the same family, a genetic variant may result in FTD, MND or ALS-FTSD phenotypes, strengthening the concept of an FTLD/MND-TDP-43 disease spectrum.
More than half of ALS-FTSD with a known genetic variant are caused by hexanucleotide repeat expansions in the C9orf72 gene [1, 2]. Variants in VCP, FUS, TARDBP, SQSTM1, OPTN, UBQLN2 and TBK1 are included in the remaining genetic spectrum [1, 2]. The TBK1 (tumor necrosis factor-associated factor NF-κB activator (TANK)-binding kinase 1) gene is located on chromosome 12 and encodes a 729-amino acid multifunctional protein kinase that acts as a regulator of various cellular pathways, including inflammation, immune response, insulin signaling, cell proliferation and autophagy [3]. Previous reported carriers of TBK1 variants are clinically heterogeneous, presenting with variable language, behavior, movement and MNDs [4, 5]. Here, we describe a case of ALS-FTSD caused by a novel TBK1 variant with an atypical phenotype.
| Case Report | ▴Top |
A 57-year-old right-handed woman presented with an 8-month history of progressive speech and memory impairments. She reported slowness of speech, wordfinding and naming difficulties, whereas comprehension, reading and writing were preserved. Other cognitive, behavioral or motor complaints were denied. She was a previously healthy operating room nurse, without usual medication or known family history of neurological disease. Neurological examination revealed a slow, telegraphic and hesitant speech, with short sentences, anomia, syllable suppression or extension, a mini-mental state examination of 27/30 (loss of 2 points on recall task and 1 point on write sentence task). Apart from hyperreflexia on the right upper limb, the remaining motor exam was normal. There were no signs of extrapyramidal, cerebellar, sensory or cranial nerves involvement. Neuropsychological assessment disclosed apraxia of speech dominant over a non-fluent language disorder, with severe alterations in verbal fluency and trail making tests, naming defect with normal performance in repetition and reading tasks, despite the increased effort to achieve them; she also had mild to moderate changes in memory tests but the pattern of performance on these tests was not suggestive of a significant defect in consolidation. Brain magnetic resonance imaging (MRI) (Fig. 1) demonstrated a left predominant mesial temporal and frontal lobe atrophy, as well as a corticospinal tract hyperintensity in T2/fluid-attenuated inversion recovery (FLAIR) and hypointensity in T2* in precentral gyri (motor band sign), most remarkable on the left (Fig. 1). Hypometabolism in the left parietal-temporal and frontal lobes was also evident in fluorodeoxyglucose-positron emission tomography (FDG-PET) (Fig. 2). Cervical MRI excluded spinal cord compression and electromyography, obtained at presentation and repeated in a late stage of disease, excluded a denervation process. Blood tests were normal and cerebrospinal fluid (CSF) biomarkers pointed to an unspecific pattern with slight elevated tau protein (446 pg/mL, normal range ≤ 335 pg/mL). Hexanucleotide repeat expansions in C9orf72 were excluded. Multigene panel sequencing (including 19 genes: CHCHD10, CHMP2B, CSF1R, FUS, GRN, HNRNPA1, HBRNPA2B1, MAPT, OPTN, PRKAR1B, SIGMAR1, SQSTM1, TARDBP,TBK1, TREM2, TUBA4A, UBQLN2 and VCP) revealed a heterozygous variant (c.1372_1373del (p.(Val458Serfs*18)) in TBK1, not previously reported in the medical literature. DNA from other family members was not available to test segregation. This variant results in a premature STOP codon leading to a truncated protein and, for that reason, it was classified as likely pathogenic.
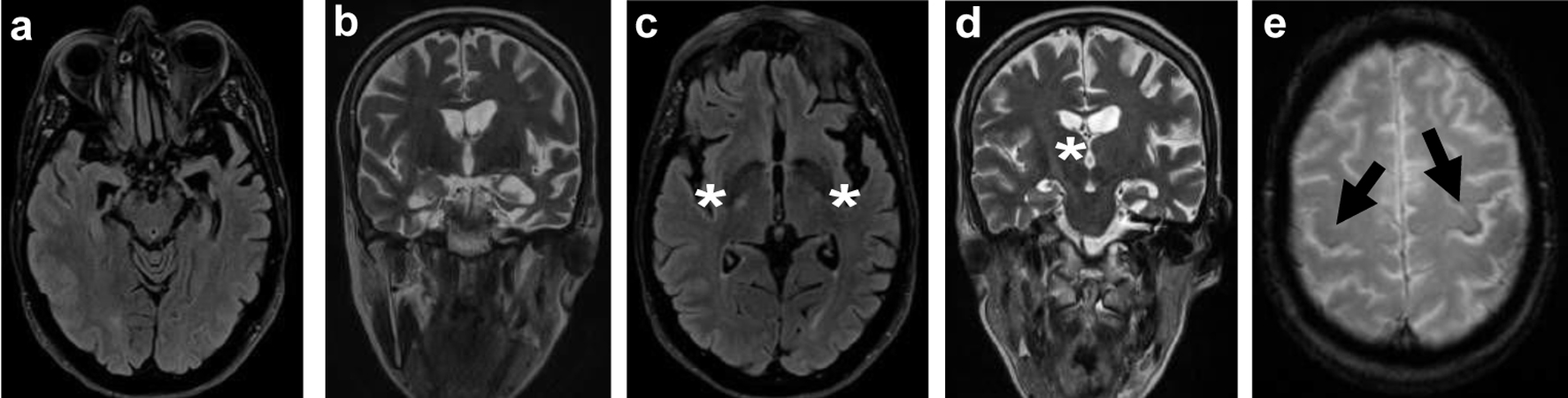 Click for large image | Figure 1. Brain MRI: axial FLAIR (a) and T2-weighted imaging (b) showing asymmetrical temporal lobe atrophy more pronounced on the left. Axial FLAIR (c) and coronal T2-weighted imaging (d) revealing hyperintensity in the posterior limb of the internal capsule and corticospinal tracts (white *). Axial T2 GRE (e) showing iron deposition (loss of signal) in the premotor cortex, most notably in the left precentral gyrus, known as the “motor band sign” (black arrows). MRI: magnetic resonance imaging; FLAIR: fluid-attenuated inversion recovery. |
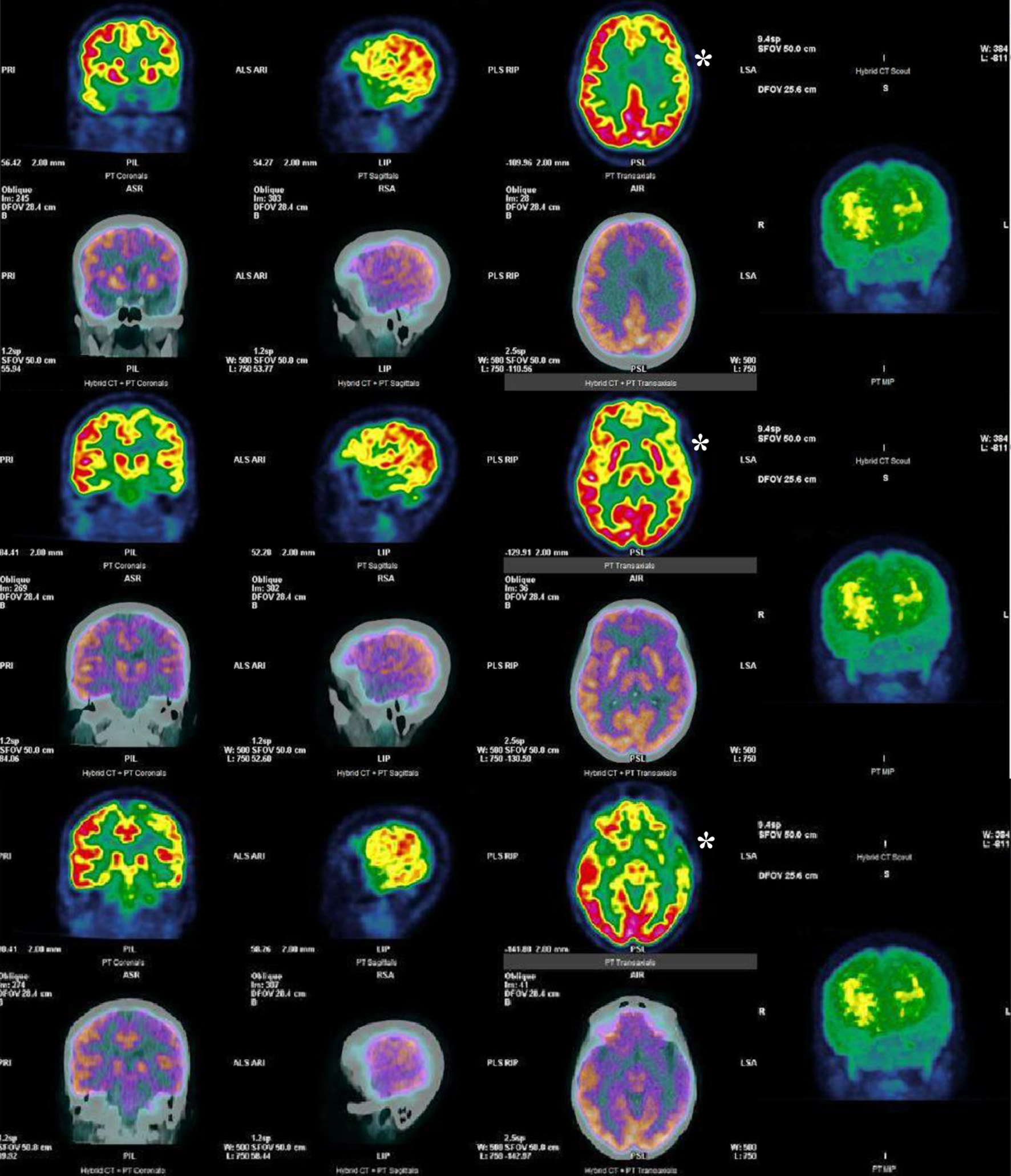 Click for large image | Figure 2. FDG-PET showing reduced [18F]FDG uptake in the left parietal-temporal and frontal lobes (white *). FDG-PET: fluorodeoxyglucose-positron emission tomography. |
Over the next 2 years, her clinical condition rapidly progressed to include pseudobulbar affect, anarthria, dysphagia, inability to protrude the tongue, dysphagia and a right predominant tetrapyramidal syndrome. Nevertheless, while it was possible to test, comprehension and writing remained unaffected. At 3 years of follow-up, she was unable to leave the bed, totally dependent on nursing care for personal care needs and a gastrostomy feeding tube was inserted to ensure nutrition. She died at the age of 61, 4 years after clinical presentation.
| Discussion | ▴Top |
Here, we have described a case of ALS-FSTD in association with a previously unreported variant in TBK1. The novelty of this report lies on the rarity of the clinical phenotype, initially presenting with apraxia of speech followed by and exclusively upper MND. Over half of patients with an ALS-FTSD present with a progressive upper and lower MND, most frequently fulfilling criteria for ALS, which is accompanied later in disease progression with behavioral disturbances in up to 75% of patients, and cognitive dysfunction (executive, memory or language deficits) in one quarter [2]. Nevertheless, our patient firstly presented with a progressive speech disorder without behavioral dysfunction or motor impairment. Apraxia of speech is thought to result from the impaired planning and programming of the sensorimotor commands necessary for directing movements that result in phonetically and prosodically normal speech. In this speech disorder, the phonetic and prosodic impairment is dominant over the language disturbance that can coexist, which is demonstrated by the patients’ retained capacity to write and understand. Apraxia of speech occurs early in various neurodegenerative disorders; therefore, long-term follow-ups watching for additional signs and symptoms are crucial to define the clinical syndrome [2]. In fact, previous reports have shown that up to a quarter of patients with FTD may develop an MND years after disease onset [6].
The absence of evidence for lower motor neuron degeneration was also noteworthy in this patient. Throughout the course of disease, she developed a tetrapyramidal and pseudobulbar syndrome compatible with primary lateral sclerosis (PLS). PLS is a rare form of MND, which only affects upper motor neurons. Both ALS and PLS have some clinical overlap with FTD but PLS is typically uncommon in FTD patients [7]. Neuroimaging studies were consistent with the diagnosis of ALS-FTSD by excluding other causes for upper motor dysfunction and showing a left predominant frontal and temporal lobe atrophy associated with indirect signs of pyramidal pathway degeneration. While these neuroimaging findings support the clinical suspicion of MND-FTSD, it can equally be observed in sporadic disease. Similar patterns have previously been reported and further explored by other researchers [8, 9]. Genetic testing should be offered for all patients with ALS-FTSD [1]. After exclusion of hexanucleotide expansion repeats in the C9orf72 gene, a new variant in TBK1 gene was found in this patient. TBK1 loss-of-function variants cause either a frameshift or a premature stop codon, leading to an absent or truncated protein product and suggesting that these variants are likely to be pathogenic through haploinsufficiency [10]. Even though we had no family history and we could not perform a segregation test of the variant, the bioinformatic analysis suggested that it has a deleterious impact and so is highly likely to be pathogenic, as it resulted in the premature inclusion of a STOP codon. Ongoing research on neurodegenerative disorders have shown that variants in TBK1 are the fourth most common cause of familial FTD and may be the second most common cause of an ALS-FTSD [11, 12]. Notwithstanding, the full clinical spectrum of TBK1 variant carriers is not completely elucidated, with nearly 100 patients reported [4]. The behavioral variant of FTD (in two-thirds of patients) and PPA (in one-quarter) are the most common clinical presentations, with (in one-quarter) or without MND [1, 2]. Literature available cases included two brothers presenting with PPA and asymmetric-onset primary lateral sclerosis [13]. On the other hand, a growing number of TBK1 variants have also been found in patients with corticobasal syndrome, progressive supranuclear palsy and progressive cerebellar ataxia, expanding the phenotypic spectrum of TBK1-associated disease [5, 14]. Blood samples from other family members, as well as a postmortem examination of brain tissue would add value to the report of this novel variant.
Conclusion
In summary, we describe a novel TBK1 variant in a patient with a rapidly progressive neurodegenerative disorder, presenting with apraxia of speech and upper MND. Following exclusion of C9orf72 gene variants, sequencing of the TBK1 gene should be considered in patients presenting with FTD, particularly with a positive family history or secondarily associated with an MND. Population-based studies are needed to clarify the prevalence and clinical phenotypes of TBK1 variant carriers.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
Informed consent for publication of this report was obtained from a patient’s relative.
Authors Contributions
All the authors had substantial contributions to conception and design, acquisition of data or analysis and interpretation of data. In the same way, all the authors participated in the drafting the article or reviewing it critically for important intellectual content. All the authors approved the final version to be published and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Data Availability
The authors declare that data supporting the findings of this article are available within the article.
| References | ▴Top |
- Strong MJ, Abrahams S, Goldstein LH, Woolley S, McLaughlin P, Snowden J, Mioshi E, et al. Amyotrophic lateral sclerosis - frontotemporal spectrum disorder (ALS-FTSD): Revised diagnostic criteria. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(3-4):153-174.
doi pubmed pmc - Burrell JR, Halliday GM, Kril JJ, Ittner LM, Gotz J, Kiernan MC, Hodges JR. The frontotemporal dementia-motor neuron disease continuum. Lancet. 2016;388(10047):919-931.
doi pubmed - Oakes JA, Davies MC, Collins MO. TBK1: a new player in ALS linking autophagy and neuroinflammation. Mol Brain. 2017;10(1):5.
doi pubmed pmc - Cui R, Tuo M, Li P, Zhou C. Association between TBK1 mutations and risk of amyotrophic lateral sclerosis/frontotemporal dementia spectrum: a meta-analysis. Neurol Sci. 2018;39(5):811-820.
doi pubmed - Wilke C, Baets J, De Bleecker JL, Deconinck T, Biskup S, Hayer SN, Zuchner S, et al. Beyond ALS and FTD: the phenotypic spectrum of TBK1 mutations includes PSP-like and cerebellar phenotypes. Neurobiol Aging. 2018;62:244.e249-244.e213.
doi pubmed - Burrell JR, Kiernan MC, Vucic S, Hodges JR. Motor neuron dysfunction in frontotemporal dementia. Brain. 2011;134(Pt 9):2582-2594.
doi pubmed - de Vries BS, Rustemeijer LMM, van der Kooi AJ, Raaphorst J, Schroder CD, Nijboer TCW, Hendrikse J, et al. A case series of PLS patients with frontotemporal dementia and overview of the literature. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(7-8):534-548.
doi pubmed - Vinceti G, Olney N, Mandelli ML, Spina S, Hubbard HI, Santos-Santos MA, Watson C, et al. Primary progressive aphasia and the FTD-MND spectrum disorders: clinical, pathological, and neuroimaging correlates. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20(3-4):146-158.
doi pubmed pmc - Long Z, Irish M, Piguet O, Kiernan MC, Hodges JR, Burrell JR. Clinical and neuroimaging investigations of language disturbance in frontotemporal dementia-motor neuron disease patients. J Neurol. 2019;266(4):921-933.
doi pubmed - Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Muller K, Marroquin N, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci. 2015;18(5):631-636.
doi pubmed - Dols-Icardo O, Garcia-Redondo A, Rojas-Garcia R, Borrego-Hernandez D, Illan-Gala I, Munoz-Blanco JL, Rabano A, et al. Analysis of known amyotrophic lateral sclerosis and frontotemporal dementia genes reveals a substantial genetic burden in patients manifesting both diseases not carrying the C9orf72 expansion mutation. J Neurol Neurosurg Psychiatry. 2018;89(2):162-168.
doi pubmed - Dong L, Wang J, Liu C, Li J, Mao C, Huang X, Chu S, et al. Genetic spectrum and clinical heterogeneity of chinese frontotemporal dementia patients: data from PUMCH dementia cohort. J Alzheimers Dis. 2022;89(3):893-901.
doi pubmed pmc - Hirsch-Reinshagen V, Alfaify OA, Hsiung GR, Pottier C, Baker M, Perkerson RB, 3rd, Rademakers R, et al. Clinicopathologic correlations in a family with a TBK1 mutation presenting as primary progressive aphasia and primary lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20(7-8):568-575.
doi pubmed pmc - Lamb R, Rohrer JD, Real R, Lubbe SJ, Waite AJ, Blake DJ, Walters RJ, et al. A novel TBK1 mutation in a family with diverse frontotemporal dementia spectrum disorders. Cold Spring Harb Mol Case Stud. 2019;5(3):a003913.
doi pubmed pmc
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Neurology Research is published by Elmer Press Inc.